
Abstract
Resolving how our brains encode information requires an understanding of the cellular processes taking place during memory formation. Since the 1970s, considerable effort has focused on determining the properties and mechanisms underlying long-term potentiation (LTP) at glutamatergic synapses and how these processes influence initiation of new memories. However, accumulating evidence suggests that long-term depression (LTD) of synaptic strength, particularly at glutamatergic synapses, is a bona fide learning and memory mechanism in the mammalian brain. The known range of mechanisms capable of inducing LTD has been extended to those including NMDAR-independent forms, neuromodulator-dependent LTD, synaptic depression following stress, and non-synaptically induced forms. The examples of LTD observed at the hippocampal CA1 synapse to date demonstrate features consistent with LTP, including homo- and heterosynaptic expression, extended duration beyond induction (several hours to weeks), and association with encoding of distinct types of memories. Canonical mechanisms through which synapses undergo LTD include activation of phosphatases, initiation of protein synthesis, and dynamic regulation of presynaptic glutamate release and/or postsynaptic glutamate receptor endocytosis. Here, we will discuss the pre- and postsynaptic changes underlying LTD, recent advances in the identification and characterization of novel mechanisms underlying LTD, and how engagement of these processes constitutes a cellular analog for the genesis of specific types of memories.
Introduction
Understanding the neural correlates of memory genesis remains a major challenge for neuroscience research. Effective treatment of memory disorders requires an intimate understanding of cellular mechanisms supporting enduring memory formation. Memories are encoded at the cellular level through changes in synaptic function resulting from neuronal activity, a process known as synaptic plasticity (Kandel 2001). Considerable evidence exists implicating synaptic plasticity as the key biological substrate for associative learning and long-term memory (Collingridge and others 2010; Ge and others 2010; Kandel 2001; Nabavi and others 2014; Whitlock and others 2006). Experiments demonstrating that synaptic strength changes in an activity-dependent manner date back to the 1970s (Bliss and Lomo 1973). These experiments showed that repeated tetanization of entorhinal cortex axons which synapse onto dentate gyrus granule cell dendrites, resulted in increased current carrying capacity of these synapses, a process now known as long-term potentiation (LTP). It is believed that this potentiation of synaptic responses acts as a cellular “engram” or trace capable of representing neuronal activity associated with memory encoding (Huganir and Nicoll 2013; Kandel 2001). Since its initial discovery, enormous effort has focused on elucidating the cellular mechanisms underlying LTP. However, data supporting a bidirectional model of synaptic plasticity in which memory formation is facilitated through both potentiation and depression of synaptic strength continues to accumulate. Long-term depression (LTD) is characterized by activity-driven, enduring reductions in synaptic efficacy (Collingridge and others 2010; Dudek and Bear 1992; Dunwiddie and Lynch 1978; Heynen and others 1996). Typically studied in the hippocampus, a brain structure essential for making new memories, LTD is gaining traction as a cellular mechanism required for certain types of memory. LTD may serve several functions including (1) increasing the dynamic range in which synapses can operate, (2) preventing synapses from entering states of saturation, (3) encoding distinct aspects of memory-inducing events, and (4) adjustment of synaptic weights to refine memory fidelity.
LTD shares several important characteristics with memory genesis such as a requirement for neuronal activity, long duration (hours in slices, days to weeks in vivo), and engagement of signaling cascades linked to memory formation (Collingridge and others 2010; see Table 1). Similar to memory and LTP, LTD requires NMDA receptors (Dudek and Bear 1992; Fox and others 2006; Liu and others 2004), calcium-dependent signaling molecules (Coultrap and others 2014; Mulkey and others 1994), and protein synthesis (Huber and others 2001; Kauderer and Kandel 2000; Mockett and others 2011; Sajikumar and Frey 2003). Treatments that impair LTD reduce memory consolidation (Ge and others 2010). Conversely, LTD expression is observed during learning, which facilitates memory acquisition (Dong and others 2012; Lemon and Manahan-Vaughan 2006; Lemon and others 2009) or reversal (Dong and others 2013; Kim and others 2011). Although much remains to be determined, considerable progress has been made in identifying cellular mechanisms underlying LTD and how LTD contributes to memory formation.
Long-Term Depression (LTD) Parallels the Fundamental Properties of Long-Term Potentiation (LTP) Associated With Memory.
The Presynaptic Side of LTD
Elucidation of the mechanisms and locus (pre- or postsynaptic) of NMDAR-dependent LTD expression lagged significantly behind its initial discovery (Dunwiddie and Lynch 1978), due to lack of reliable induction protocols. This was remedied in part through the introduction of a prolonged, low-frequency stimulation protocol (900 pulses, 1 Hz; Dudek and Bear 1992), which yields more consistent LTD in young animals. However, doubts remained about the mechanisms through which LTD was being induced—was it similar to LTP, through NMDAR activation, or did it require metabotropic glutamate receptor (mGluR) stimulation (Bashir and others 1993)? Subsequent experiments demonstrated that both mGluRs (using the group 1 mGluR agonist, (RS)-3,5-dihydroxyphenylglycine [DHPG], Palmer and others 1997, or low-frequency paired pulse protocols, Kemp and others 2000) and NMDARs (prolonged LFS; Dudek and Bear 1992) were capable of inducing LTD. Although both forms of LTD result in down-regulation of synaptic transmission and demonstrate overlap in some mechanisms of expression, divergence exists in several aspects of these forms of LTD. For instance, whereas NMDAR-LTD demonstrates developmental differences with ease of induction being initially high when young and becoming more difficult with age, mGluR LTD can readily be induced independent of age (Kemp and others 2000).
Generally, the primary mechanism for decreasing synaptic transmission presynaptically is through changes in the probability of vesicle release. Initial evidence using FM1–43 dyes to quantify vesicular turnover rates suggested a reduction in release rates following the induction of LTD (Stanton and others 2003). This presynaptic effect was dependent on postsynaptically released nitric oxide (NO), which through retrograde transmission enhanced presynaptic cGMP resulting in modulation of vesicle kinetics (Stanton and others 2003). However, the role of NO remains controversial and poorly understood as NO synthase inhibitors failed to prevent NMDAR-LTD (Cummings and others 1994; Malen and Chapman 1997), suggesting that further experiments are required to determine the precise role of NO in this form of LTD. Corroborating evidence for a change in presynaptic release probability was provided using dual-photon laser-scanning microscopy of FM1–43 while varying extracellular calcium concentration to facilitate visualization of the readily releasable pool (RRP) of synaptic vesicles in CA1. Vesicular release from the RRP was persistently reduced following NMDA-LTD (Zhang and others 2006). Use of the nitric oxide synthase inhibitor N-omega-nitro-L-arginine attenuated the induction of NMDA-LTD, suggesting that retrograde transmission through NO constitutes part of the mechanism responsible for readily releasable pool down-regulation (Zhang and others 2006).
To more accurately measure changes in neurotransmitter release dynamics during LTD, Xu and others (2013) developed a method that allowed them to label vesicular glutamate transporters with biotin during neurotransmitter release. Pairing of vesicle glutamate transporter tagging with GluA1 AMPAR subunit biotinylation allowed for simultaneous measurement of pre- and postsynaptic activity and sensitivity, respectively. Application of this method to cultured rat hippocampal neurons revealed that presynaptic glutamate release actually increases during mGluR-LTD as determined by quantifying the amount of presynaptic VGluT1 biotinylation observed following treatment with the mGluR agonist DHPG (Xu and others 2013). This enhanced presynaptic release was preceded by decreases in postsynaptic mEPSC amplitude consistent with a homeostatic up-regulation of glutamate release in response to decreased levels of postsynaptic AMPARs. Further evidence for a homeostatic up-regulation of glutamate release was provided by inhibiting GluA2 endocytosis with the Tat-GluR23y peptide, which blocked both LTD and the increases in presynaptic activity observed following DHPG treatment (Xu and others 2013). This study demonstrated through comonitoring of pre- and postsynaptic activity levels that mGluR-LTD induction is a dynamic process in which decreases in postsynaptic sensitivity as a result of loss of synaptic AMPARs is partially compensated for by increased glutamate vesicle turnover during LTD. It will be of interest to characterize the transsynaptic interactions that drive this homeostatic compensation to determine how modulation of pre- and postsynaptic components of synaptic transmission coordinate to regulate changes in synaptic strength.
Postsynaptic Mechanisms Mediating NMDAR-LTD
A requirement for NMDAR activation suggests a dependence on intracellular calcium for the induction of prolonged, LFS-induced LTD in the postsynaptic neurons. This was confirmed through manipulations that targeted intra- and extracellular calcium levels (Mulkey and Malenka 1992). Although disagreement continues regarding the contribution of specific GluN subunits to LTD, a dependence on the GluN2B subunit of the NMDAR has been linked to LTD (Brigman and others 2010; Ge and others 2010; Li and others 2006; but see Li and others 2007). Brain slice orientation influences the recruitment of specific receptor subtypes during LTD; sagittal slices demonstrated LTD that preferentially engaged signaling cascades requiring muscarinic receptor activation, whereas coronal sections yielded prototypical LTD dependent more so on GluN2B subunits. Blocking muscarinic receptors in sagittal slices shifted the dependence of LTD back to the GluN2B-dependent form (Bartlett and others 2011). Corroborating data generated in mice in which GluN2B is post-developmentally and selectively knocked out of CA1 pyramidal cells demonstrated impaired NMDAR-dependent LTD induced by low-frequency stimulation combined with glutamate transporter inhibition (Brigman and others 2010).
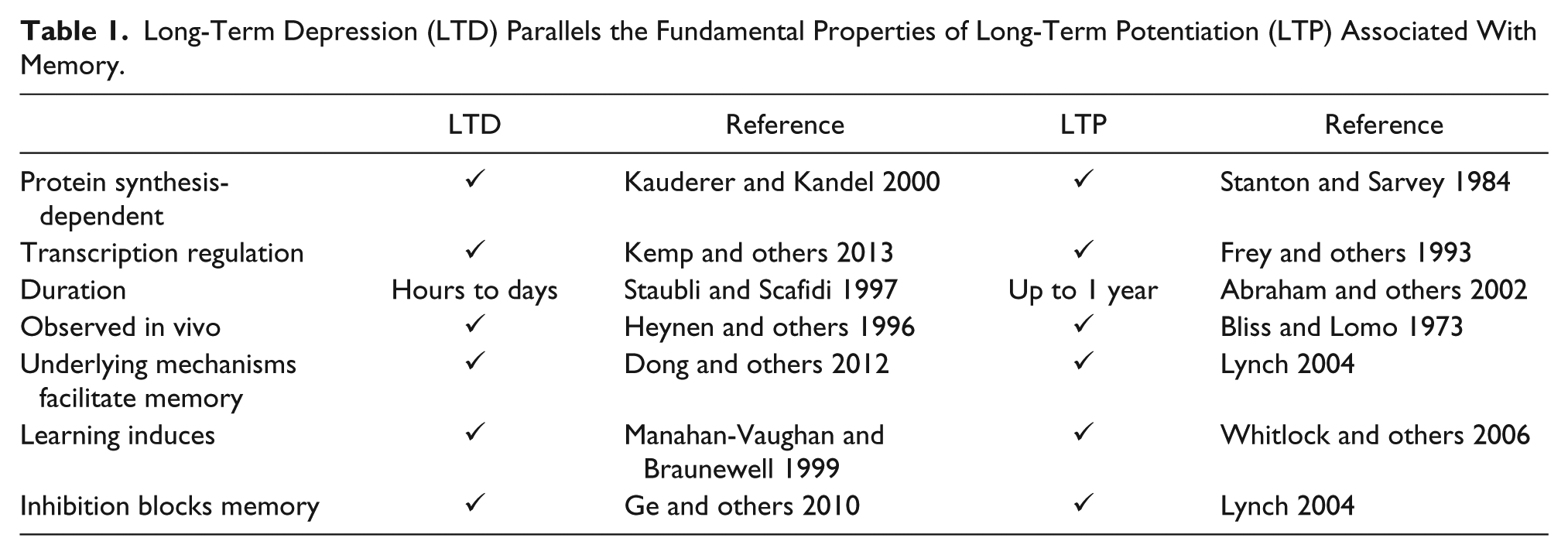
Considerable evidence characterizing the calcium-dependent, downstream signaling cascades initiated following NMDAR activation has revealed several key signaling molecules that regulate LTD. Originally proposed by Lisman (1989) was the concept of differential sensitivity of phosphatases and kinases to calcium levels, with phosphatases demonstrating higher affinity relative to kinases. This concept provided an elegant explanation for the discrepancy in frequency dependence of LTP and LTD; high-frequency (100 Hz) tetanization generates large, transient increases in calcium that preferentially engage LTP-inducing kinases such as the Ca2+-dependent, Ca2+/calmodulin-dependent kinase II (CaMKII), whereas LFS results in lower levels of calcium influx over an extended time scale resulting in selective activation of LTD-producing phosphatases such as calcineurin (Ca2+-dependent phosphatase, also known as protein phosphatase 2B [PP2B]). LTD induced via LFS produces a transient increase in phosphatase activation through Ca2+-dependent initiation of calcineurin. Calcineurin dephosphorylates and inactivates the protein phospatase-1 (PP1) inhibitor, inhibitor-1 (I-1). Once removed from I-1 repression, PP1 dephosphorylates AMPARs resulting in their internalization and depression of synaptic strength (Mulkey and others 1993, 1994) (Fig. 1). PP1 dephosphorylates AMPARs at site Ser845, thereby facilitating the endocytosis and synaptic removal of these receptors (Kameyama and others 1998; Lee and others 2000). Thus, phosphatases facilitate LTD through dephosphorylation of AMPARs, which decreases AMPAR currents and increases AMPAR endocytosis. Synaptic localization of calcineurin appears crucial for LTD, as demonstrated by knock-in mice lacking the A kinase anchoring protein (AKAP) anchoring motif required for sequestering calcineurin directly at synapses, which exhibited dramatically reduced NMDAR-LTD (Sanderson and others 2012).
Canonical and novel NMDAR-dependent LTD signaling cascades. (A) Calcium influx via NMDARs activates calcineurin. (B) Calcineurin (CaN) translocates to AKAP79/150 where it dephosphorylates protein phosphatase one (PP1), which releases Inhibitor-1 (I-1) from PP1-mediated tonic suppression. Free I-1 dephosphorylates AMPARs at Ser845, which decreases AMPAR stability at the postsynaptic membrane. (C) CaMKII, although traditionally linked to LTP, can also facilitate LTD through phosphorylation site-specific regulation of AMPARs. Synaptic activity triggers activation of NMDARs and subsequent autophosphorylation and activation of CaMKII. (D) Once activated, CaMKII phosphorylates Ser567, which is associated with reductions in GluA1 synaptic localization. Note that S567 is located in the first intracellular loop, between the first two transmembrane domains of the GluA1 subunits and not on the C-terminal tail as depicted here.
A requirement for Ca2+ for both LTP (Lisman and others 2012; Morris and others 1986) and LTD (Dudek and Bear 1992; Mulkey and Malenka 1992) served as a point of contention in the synaptic plasticity field given that these forms of plasticity were initially viewed as serving diametrically opposed functions during memory formation. How could Ca2+ facilitate both LTP and LTD without masking the induction of both? Further molecular dissection revealed preferential engagement of kinases during LTP and phosphatases during LTD that partially reconciled how opposing forms of plasticity could nevertheless both require the same ionic trigger (Collingridge and others 2010). However, conflicting results regarding the role of the Ca2+-dependent kinase, CaMKII in LTD convoluted the known mechanism through which Ca2+ initiated synaptic depression (Mockett and others 2011; Schnabel and others 1999). Early studies found that mGluR-dependent LTD is enhanced by the CaMKII inhibitor, KN62 (Schnabel and others 1999), whereas more recent findings indicate CaMKII signaling enhances protein synthesis required for expression of mGluR-LTD (Mockett and others 2011). Recent data have demonstrated how CaMKII can simultaneously mediate both LTP and LTD through phosphorylation site-specific regulation of AMPAR surface expression. LTD stimulation applied either in the presence of the specific CaMKII inhibitor peptide, tatCN21, or in mice harboring a forebrain specific knockout of αCaMKII demonstrated impaired LTD (Coultrap and others 2014). The authors demonstrated that T286 autophosphorylation of CaMKII is increased during LTD and activated CaMKII phosphorylates S567 on GluA1. S567 phosphorylation has been linked to reductions in GluA1 synaptic localization and synaptic strength (Lu and others 2010) (Fig. 1). Rendering CaMKII “autonomous” through pre-phosphorylation of T286 paired with additional Ca2+/CaM stimulation revealed a saturation of S567 phosphorylation at minimal levels of autonomous T286, whereas S831 phosphorylation, which is typically observed during LTP, was increased fivefold by additional Ca2+/CaM (Coultrap and others 2014). Thus, the preferential initiation of LTP over LTD can be explained by the near maximal phosphorylation of S567 induced at lower levels of autonomous CaMKII activity. Prolonged LFS typically used to induce LTD generates near maximal phosphorylation of S567 at low levels of autonomous CaMKII activity, whereas S831 phosphorylation requires much higher levels of constitutively active CaMKII to drive synaptic expression of AMPARs and LTP. Further research is required to determine if differential sensitivity of S567 and S831 (or S845) to phosphatases plays a role in tipping the balance of plasticity toward LTP or LTD.
Regulation of AMPAR Function and Localization during LTD
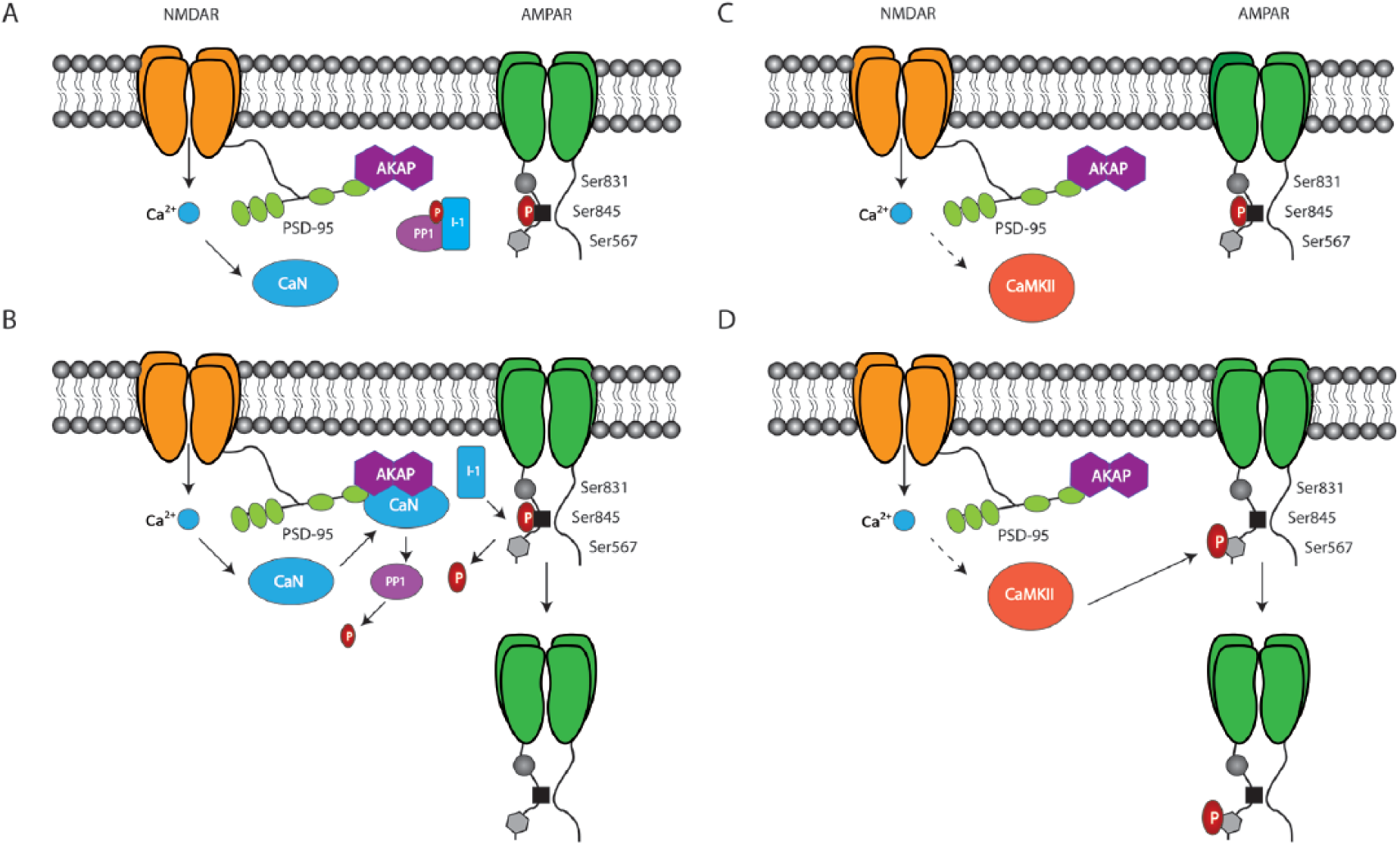
A postulated mechanism for mediating long-term decreases in synaptic strength was to decrease AMPAR surface expression through regulation of endocytosis. Confirmation of AMPAR internalization associated with NMDAR-LTD was achieved using antibodies that recognize extracellular regions of AMPARs in cultured neurons (Beattie and others 2000). Although evidence exists suggesting GluA1 dephosphorylation of Ser845 results in LTD (Kameyama and others 1998), it has previously been established that clathrin-dependent endocytosis is mediated primarily through the GluA2 subunit of AMPARs (Man and others 2000; Wang and Linden 2000; however, see Meng and others 2003 for evidence that GluA2 is not required for LTD and Granger and Nicoll 2014 who showed that glutamatergic receptors can interchangeably support LTD regardless of subtype). These groups showed increased internalization of HA-tagged GluA2 in response to LTD-inducing stimuli, which could be blocked by inhibiting clathrin-mediated endocytosis (Man and others 2000; Wang and Linden 2000). Additionally, proteins that share binding sites on GluA2 including N-ethylmaleimide-sensitive factor, NSF, and the clathrin adaptor protein, AP-2, control AMPAR cycling with NSF promoting stabilization of synaptically localized AMPARs and AP-2 promoting AMPAR endocytosis (Lee and others 2002). Blocking the interaction of NSF with GluA2 occludes NMDAR-LTD (Lüscher and others 1999), and a blocking peptide that prevents interaction of AP-2 with GluA2 inhibits the induction of LTD and LTD-dependent spatial memory formation (Dong and others 2013; Ge and others 2010). An emerging player in the bidirectional regulation of AMPARs surface expression through NSF and AP-2 is the neuronal calcium sensor, hippocalcin.
Hippocalcin translocates and binds to the plasma membrane in the presence of calcium, where it interacts with both AP-2 and GluA2 and promotes the exchange of NSF for AP-2 during LTD (Palmer and others 1997; see Fig. 2). A recently identified alternative signaling pathway connecting LTD to GluA2 interacting proteins was shown to be mediated through calcineurin and dephosphorylation of phosphatidylinositol 4-phosphate 5-kinase γ661 (PIP5Kγ661). Dephosphorylated PIP5Kγ661 becomes active on associating with AP-2 and produces PI(4,5)P2, driving further sequestration of AP-2 and other cellular machinery required for endocytosis. Postsynaptic overexpression of a kinase-dead PIP5Kγ661 mutant completely attenuated LFS-induced LTD (Unoki and others 2012). Taken together these data suggest that regulation of GluA2 interacting proteins serves as a highly dynamic method through which AMPAR endocytosis is coordinated during LTD.
Parallel mechanisms converge on NSF/AP2 to regulate GluA2-dependent LTD. (A) Under basal conditions, N-ethylmalemide-sensitive factor (NSF) binds to the GluA2 subunit, which maintains AMPAR stability at the postsynaptic membrane. (B) Activation of the calcium sensor protein, hippocalcin (Hippo), induces its translocation and binding to the synaptic membrane where it promotes AP-2 exchange for NSF on GluA2. AP-2 triggers clathrin-dependent endocytosis of AMPARs required for LTD. (C) The phosphatidylinositol kinase, PIPKγ661 is inactive when phosphorylated. (D) CaN dephosphorylates PIPKγ661, which upon associating with AP-2 becomes fully activated. PIPKγ661-AP-2 binding promotes further production of AP-2, which sequesters cellular products required for endocytosis in addition to displacing NSF.
An alternative pathway for tonically suppressing AMPAR expression independent of degradation is trapping AMPARs extrasynaptically. Following endocytosis, AMPARs enter complex trafficking pathways that determine the extent of LTD. Targeting of internalized AMPARS to divergent post-endocytotic pathways is mediated through several mechanisms. When AMPARs enter Rab11-dependent recycling endosomes LTD is suppressed as AMPARs are trafficked back to the postsynaptic membrane (Fernández-Monreal and others 2012). Conversely, other internalized AMPARs are trafficked via Rab7-dependent late endosomes toward lysosomes for degradation. However, inhibition of lysosomal function does not affect synaptic depression, suggesting that transport of AMPARs through Rab7 facilitates LTD independent of degradation (Fernández-Monreal and others 2012).
Endocytosis of AMPARs is followed by persistent sequestration of AMPARs to non-synaptic pools (Citri and others 2010). When its activation is suppressed, protein interacting with C kinase 1 (PICK1) associates with molecular complexes capable of sequestering AMPARs during LTD (Jin and others 2006; Kim and others 2001; Terashima and others 2008). PICK1 through its N-terminal PSD-95/Dlg/ZO1 (PDZ) domain directly interacts with AMPARs containing subunits GluA2/3 (Staudinger and others 1997), as well as with the glutamate receptor interacting protein, GRIP1 (Jin and others 2006; Lu and Ziff 2005). The combination of PICK1’s binding partners and its calcium sensitivity (Hanley and Henley 2005) confers PICK1 with the capacity for regulating Ca2+-dependent AMPAR trafficking. Lentivirus-mediated molecular replacement was used to determine that PICK1 activity is prevented during NMDAR-dependent LTD in a calcium-dependent manner, which promotes its association with and sequestration of endocytosed AMPARs, thus preventing their recycling back to synapses (Citri and others 2010).
Recent Advances in the Molecular Dissection of NMDAR-LTD
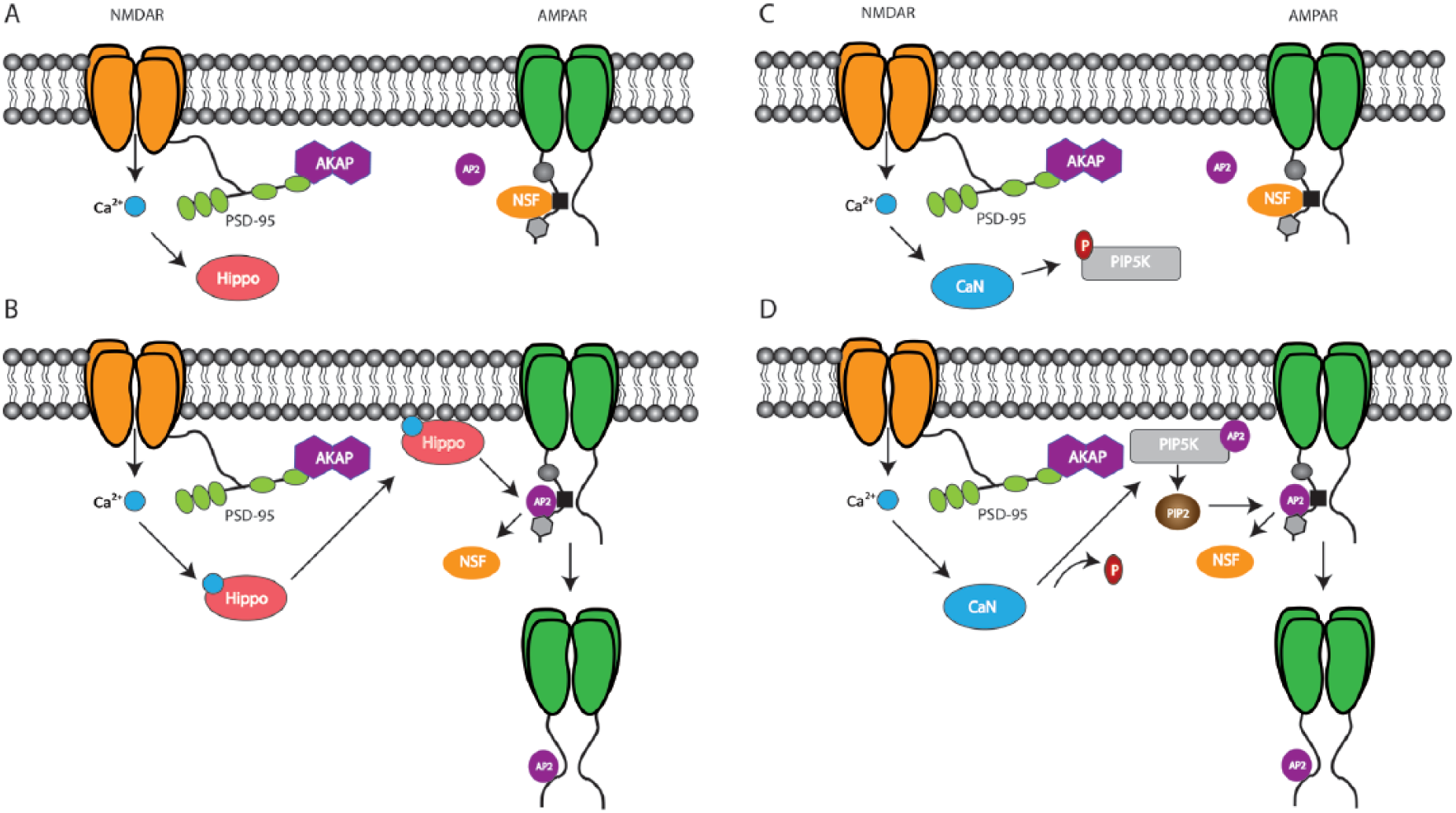
At the molecular level, the list of known Ser/Thr kinases that mediate NMDAR-LTD continues to grow. Original work demonstrating suppression of LTP by the Ser/Thr kinase, GSK-3β (Glycogen Synthase Kinase-3; Peineau and others 2007) has been bolstered through data demonstrating how GSK-3β regulates PSD-95 localization to enhance LTD. GSK-3β phosphorylates PSD-95 on Threonine 19 (T19) in cultured hippocampal cells treated with NMDA to induce LTD (Nelson and others 2013). PSD-95 synaptic clustering potentiates synaptic transmission which T19 phosphorylation opposes through driving PSD-95 dispersal and subsequent AMPAR internalization during LTD (Nelson and others 2013) (Fig. 3). Thus, GSK-3β appears to be a key regulator of NMDAR-LTD although further in vivo validation is required to fully characterize its role in LTD-dependent memory processes.
GSK-3β enhances LTD through dispersal of PSD-95. (A) Auxiliary proteins such as TARP facilitate AMPAR stabilization through interaction with the MAGUK protein, PSD-95. Phosphorylation suppresses GSK-3β activity preventing it from interacting with PSD-95. (B) NMDAR stimulation initiates GSK-3β which phosphorylates PSD-95 at T19 which drives PSD-95 dispersal and subsequent internalization of AMPARs during LTD.
A role for tyrosine phosphorylation in NMDAR-LTD has previously been established (Ahmadian and others 2004; Hayashi and Huganir 2004). Evidence characterizing how tyrosine dephosphorylation may trigger LTD has implicated the GTPase Arf6 guanine nucleotide exchange factor, BRAG2. Scholz and others (2010) showed that BRAG2 interacts with tyrosine 876 (Y876) in the C-terminal of GluA2 and binding of GluA2 to BRAG2 causes an increase in BRAG2 catalytic activity on Arf6. The phosphorylation state of Y876 regulates BRAG2 catalytic activity with dephosphorylation increasing BRAG2 activity, whereas phosphorylation did not. Arf6 recruits AP2 to synaptic membranes thereby initiating endocytotic vesicle formation required for LTD induction (Scholz and others 2010). Thus, interfering with BRAG2 binding to GluA2 should prevent AMPAR endocytosis and LTD expression. A peptide targeting this BRAG2-GluA2 interaction (Tat-GluR23y) has successfully been used to block LTD-dependent memory formation (Dong and others 2013) (Fig. 4). Additional data supporting a role for tyrosine phosphorylation in LTD have shown that the tyrosine kinase Janus kinase 2 (JAK2) is required for the induction of NMDAR-LTD (Nicolas and others 2012). The non-receptor protein tyrosine kinase family Janus Kinases (JAKs) are activated following NMDAR stimulation and subsequently engage downstream effectors including the transcription factor, Signal Transducer and Activator of Transcription (STAT). This JAK/STAT pathway is initiated during NMDAR-LTD as pharmacological inhibition or knockdown of either JAK2 or STAT3 prevents expression of NMDAR-LTD (Nicolas and others 2012). A lack of effects of these inhibitors on depotentiation or mGluR-LTD was also observed, suggesting a mechanistic divergence between these forms of plasticity. Whole cell recordings using pipettes loaded with JAK and STAT inhibitors confirmed that the locus of LTD suppression was postsynaptic. Reconciling these new findings with previously established roles for phosphatase activation downstream of NMDARs was the finding that JAK2 activation requires PP1 and calcineurin as application of okadaic acid or cyclosporine A, respectively, decreased JAK2 activation and blocked LTD following low-frequency stimulation (Nicolas and others 2012). Specifically how the JAK/STAT pathway regulates LTD remains to be determined, but it appears to be independent of transcription regulation as preventing the JAK2-induced nuclear translocation of STAT3 did not alter LTD (Nicolas and others 2012). As the JAK/STAT pathway has been linked to Alzheimer’s disease and memory (Chiba and others 2009a, 2009b), determination of its expression in vivo should provide further insight into the roles of LTD in neuropsychiatric disease and mnemonic function.
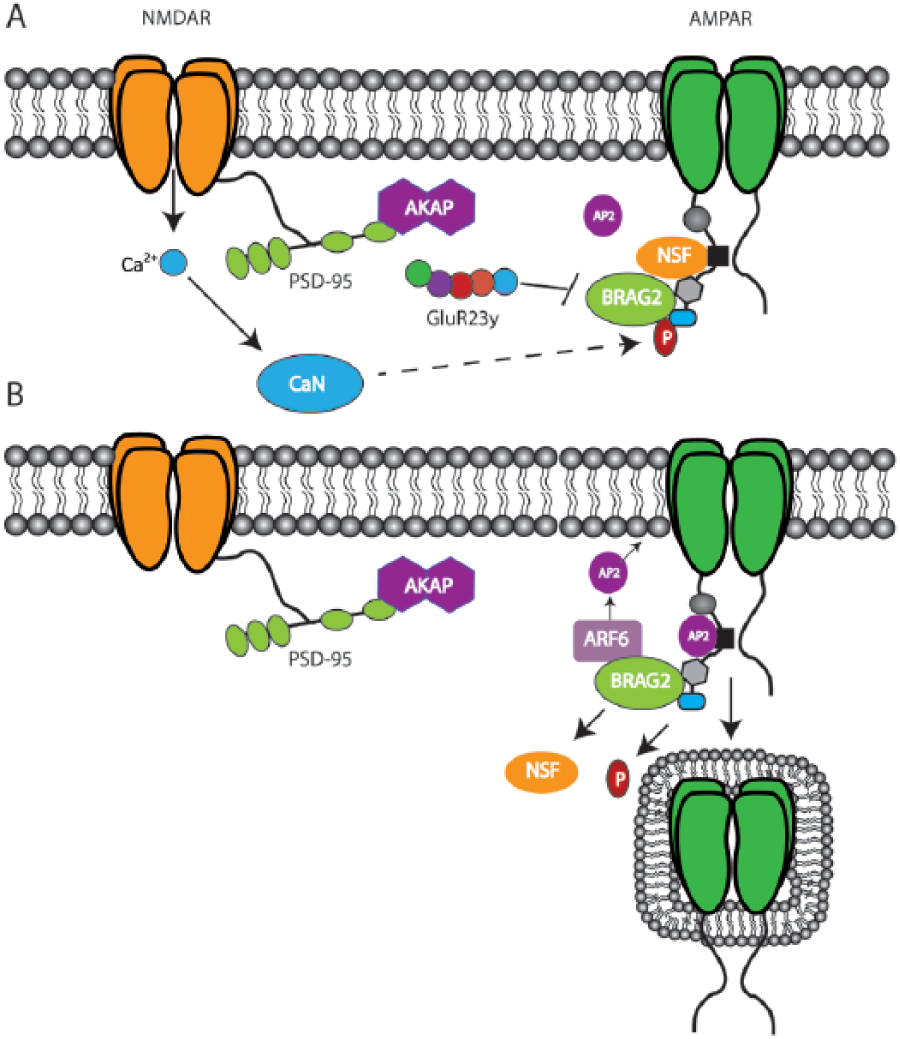
BRAG2/GluA2 interactions regulate endocytosis and can be targeted to block LTD. (A) The Arf6 GTPase, BRAG2, binds to GluA2 at Y876. Phosphorylation of Y876 represses BRAG2 activity. Endocytosis can be blocked by preventing the interaction between BRAG2 and GluA2 using the interference peptide, TAT-GluR23y. (B) Dephosphorylation of Y872 increases BRAG2 activity, which drives AP-2 recruitment by Arf-6 to the synaptic membrane. AP-2 engages endocytotic vesicle formation, which enhances AMPAR internalization and LTD expression.
An alternative form of LTD dependent on O-GlcNAcylation of AMPARs yet independent of PKC and NMDARs has recently been described. AMPAR serine residues can be modified via phosphorylation and similarly through O-linked addition of β-N-acetylglucosamine. To probe the role of AMPAR glycosylation, Taylor and others (2014) manipulated the O-GlcNAcylation of GluA2. Transiently increasing O-GlcNAcylation through treating hippocampal slices with glucosamine or inhibiting the de-glycosylating enzyme, O-GlcNacase, induced LTD (Taylor and others 2014). Co-application of APV failed to block LTD induced by application of O-GlcNacase, suggesting NMDARs were not required. However, saturation of NMDAR-dependent LTD occluded LTD requiring O-GlcNAcylation. Reversing this paradigm (saturating O-GlcNAcylation first then attempting low-frequency LTD) failed to show full occlusion. Increased GluA2 but not GluA1 O-GlcNAcylation and direct association of GluA2 with OGT, which catalyzes O-GlcNAc addition to serine/threonine residues, were also observed using co-IP, suggestive of GluA2-specific regulation (Taylor and others 2014). Taken together, these data suggest that there is partial overlap in the mechanisms mediating the expression of these forms of LTD. This may account for the observation that novel-object recognition (NOR) but not contextual fear conditioning (CFC), which both require hippocampal plasticity are differentially affected by O-GlcNAcylation, with NOR but not CFC being compromised by increased O-GlcNAcylation (Taylor and others 2014). This suggests that mechanistically divergent forms of LTD may facilitate unique types of memories with glycosylation mediating novel object memory performance and NMDAR-LTD affecting encoding of context.
What LTD Does: Putative Roles for LTD in Memory Consolidation and Memory Flexibility
The hippocampus is a bilateral brain structure that is crucially involved in several forms of memory (Anderson and others 2007). Preliminary experiments attempting to elucidate the cellular mechanisms underlying memory formation in the hippocampus were crude in nature due to the use of broad-spectrum antagonists. With the continued refinement and development of pharmacological and genetic tools capable of specifically inhibiting different components of plasticity, evidence supporting a role for LTD in various aspects of hippocampus-dependent memory formation continues to accumulate. The NMDAR was shown to be crucial for spatial memory formation as indicated by reduced performance in the Morris water maze (MWM) following injection of the NMDA receptor antagonist, AP-5 (Morris and others 1986). However, whether NMDARs were contributing to spatial memory through LTP or LTD-like mechanisms was not determined. With the combination of targeted genetic manipulations and advances in NMDAR subunit antagonist selectivity has come evidence supporting a role for LTD in memory formation. Strong evidence linking LTD to memory encoding was demonstrated through selective genetic reduction of GluN2B subunit levels in CA1 pyramidal cells. Both NMDAR-LTD and hippocampus-dependent memory were compromised following NR2B knockout (Brigman and others 2010). Specifically, this study showed that the selective ablation of GluN2B subunits in pyramidal neurons in CA1 and cortex impairs CA1 NMDAR-LTD and results in deficits in several hippocampus-dependent learning and memory tasks (Brigman and others 2010). Blocking LTD induction using a GluN2B-specific antagonist (Ro25-6981) or preventing LTD expression with inhibition of GluA2-dependent endocytosis using the TatGluR23y interference peptide before training, while having little effect on spatial learning acquisition, similarly prevented spatial memory consolidation (Ge and others 2010). Targeting intracellular signaling cascades downstream of NR2B can likewise impair learning and memory. Genetically reducing activity of a key phosphatase normally activated during LTD, calcineurin (PP2B), abolished LTD and disrupted episodic memory formation (Zeng and others 2001). Taken together, these data imply that production of LTD during the process of learning a new task may have an important function in facilitating the memory consolidation of newly learned information by suppressing any potential interference from trace memories established by previous learning in the same neuronal circuits. Consistent with this conjecture, it has been established that novel environment exploration (in which animals encode a new environment) can facilitate the production of LTD (Manahan-Vaughan and Braunewell 1999) and also the conversion of short-term memory to long-term memory (Moncada and Viola 2007). Further data from in vivo studies have shown facilitation of LTD by novelty (Manahan-Vaughan and Braunewell 1999) and spatial exploration (Lemon and Manahan-Vaughan 2012) and a requirement for LTD in novelty acquisition (Lemon and Manahan-Vaughan 2006). Similar work from this group found that novel spatial learning triggered LTD in mouse hippocampus (Goh and Manahan-Vaughan 2013). Recent experiments have extended these findings by demonstrating a requirement for LTD-like processes in the consolidation of spatial memory (Ge and others 2010).
Memory Flexibility
Flexibility in neuronal circuits is crucial for updating or unlearning irrelevant or erroneous information. LTD may facilitate this process by weakening unused synapses or facilitating synaptic depression of previously potentiated synapses. Reversal learning is used to gauge how well animals are able to adapt to changing environmental contingencies once a behavioral pattern reflecting memorization has been established. Rats subjected to a spatial reversal learning task, in which they are forced to learn the new location of a platform moved from a previously memorized quadrant showed dramatic deficits in new learning when injected with the GluN2B inhibitory, Ro25-6981 1 hour prior to the first trial on each reversal learning day. Similar results were observed when AMPAR endocytosis was specifically inhibited using the Tat-GluR23y peptide (Dong and others 2013) (Fig. 5). Inhibiting protein phosphatase 2A reduced the expression of LTD and impaired reversal learning in the MWM, suggesting that LTD may facilitate memory by enhancing synaptic flexibility required for encoding changes in environmental contingencies (Nicholls and others 2008). Knockout of the phosphatidylinositol 3-kinase (PI3K) p110γ catalytic subunits (Pik3cg−/−), which when dimerized constitute the PI3K isoform, PI3Kγ, disrupted NMDAR-LTD and significantly impaired flexibility performance when the location of a MWM platform was changed after having learned a previous location (Kim and others 2011). Taken together, these data suggest that although acquisition of a spatial memory was intact, an inability to express LTD prevents behavioral flexibility required for reversal learning.
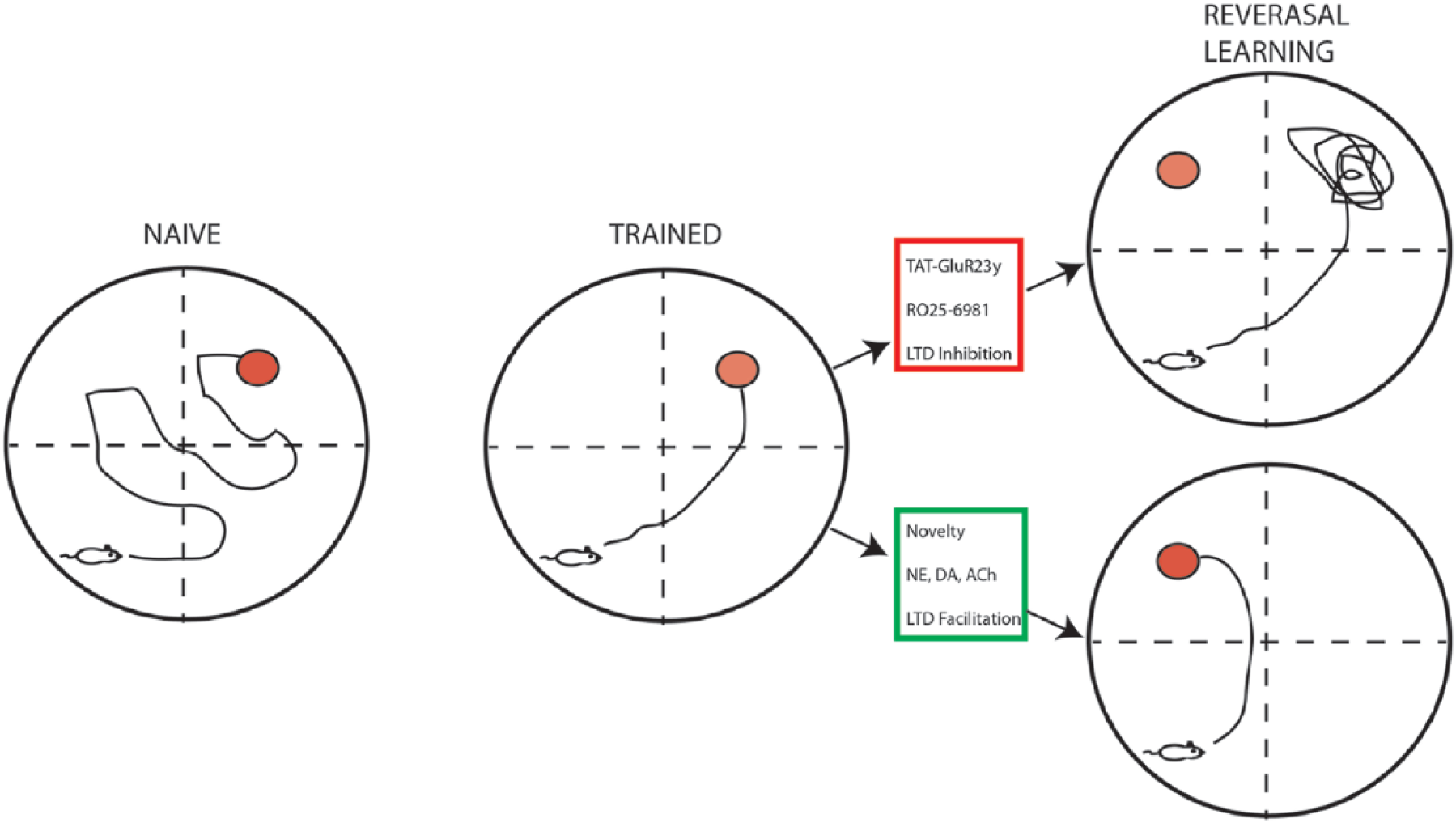
LTD facilitates memory flexibility. Naïve rodents (left) placed in the Morris water maze (MWM) will initially explore throughout the maze until they locate the escape platform (red circle). With training (middle), their swimming distance and latency to find the platform are reduced, as they form spatial memories based on visual cues located in the testing room. Shifting the platform to a new quadrant forces the rodents to relearn the task, which requires cognitive flexibility. Upper right: Blocking LTD by inhibiting endocytosis (TAT-GluR23y peptide) or NR2B receptors (RO25-6981) prevents learning of the new platform location. Bottom right: Exposure to novelty or increasing levels of pro-LTD neuromodulators facilitates learning of the new platform location, consistent with a role for LTD in memory flexibility.
Memory Suppression
The memory suppressing effects of stress can be alleviated through blocking LTD (see Howland and Wang 2008 for review). A low-frequency stimulus applied to the SC-CA1 pathway that was subthreshold for LTD could reliably induce LTD in animals subjected to 30-minute exposure on an elevated platform, constituting an acute stress (Wong and others 2007). This stress-enabled LTD was abolished by the specific NR2B antagonist Ro25-6981 or direct inhibition of LTD through prior injection of the Tat-GluR23Y peptide (Wong and others 2007). These data indicate that stress impairs memory through mechanisms requiring LTD.
Conclusions
Emerging evidence suggests that, similar to LTP, LTD is required for the instantiation of memory (Dong and others 2013; Lemon and others 2009). Our understanding of mechanisms underlying the induction and maintenance of LTD has been significantly improved, particularly during the last two decades although the mechanisms mediating the tonic suppression of AMPAR function and expression during the maintenance of LTD have yet to be fully elucidated (see Collingridge and others 2010 for review). As memories can be encoded in single trials and calcium transients are large but brief (millisecond time scale), the question remained as to how memories could endure beyond the stimulus that triggered their formation. Investigations into the protein synthesis dependence of NMDAR-LTD are beginning to yield results both in terms of the localization of the translation regulation and identification of the proteins that are being synthesized. The expression of NMDAR-LTD is inhibited by translation repressors in vitro (Huber and others 2001) and in vivo (Manahan-Vaughan and others 1999). However, evidence suggests that processes independent of macromolecule synthesis can be sufficient for maintaining memories. Initially considered the “memory molecule,” constitutively activated CaMKII was believed to be the mechanism through which transient synaptic activation could nevertheless promote enduring memory formation (Mayford and others 1996). Based on recent findings showing that CaMKII is required for both LTD and LTP (Coultrap and others 2014), one could speculate that tonic, lower level activation of CaMKII could fulfil the requirement for tonic suppression of AMPAR expression necessary for LTD-dependent memory maintenance. Alternatively, data suggesting that suppression of Protein kinase M zeta (PKMζ) activation (Hrabetova and Sacktor 1996) is associated with depotentiation, an AMPAR endocytosis-dependent process similar to LTD, suggests the possibility that this could also serve as a mechanism for enhancing LTD. It has yet to be determined if LTD-dependent memory is similarly mediated through constitutive activation or repression of a single “memory molecule,” although the likelihood of this being the case is small given the number of alternative signaling cascades and molecular players engaged during LTD. It remains to be determined precisely what intracellular signaling cascades or molecules are activated that maintain synapses in a tonically depressed state during memory genesis and to what degree these differ dependent on the type of memory being encoded.
Given the evidence for synaptic pathology including altered synapse density and function in Alzheimer’s disease patients and models (Scheff and Price 2006; Shankar and others 2008), elucidating the function and mechanisms of LTD in cognition is crucial for developing targeted therapeutics going forward. As data showing improved cognitive functioning following transcranial magnetic stimulation–based potentiation of cortical and subcortical synaptic pathways shows promising results (see Nardone and others 2011 for review), it will be interesting to determine if stimulating brain areas with LTD-like protocols will have similar therapeutic value in the treatment of disorders characterized by runaway synaptic transmission including addiction, epilepsy, or schizophrenia, a disease characterized by impaired behavioral flexibility (Floresco and others 2009). One can readily envision situations in which depressing overactive synaptic circuits, such as those observed in these pathological conditions, will serve to reestablish synaptic equilibriums, thus restoring behavioral function to near-optimal states. Coupling noninvasive neural stimulation with pharmacological treatments that likewise specifically target LTD like processes should yield therapeutic benefits to neuropsychiatric patients across a broad spectrum of disorders. Clearly, a full proteomic and physiological profile encompassing different induction protocols as well as characterizing temporal properties is required to fully understand how synaptic regulation during LTD maintenance contributes to the expression of synaptic changes required for LTD-dependent memory.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.