
Abstract
Tauopathies encompass a broad range of neurodegenerative diseases featuring extensive neuronal death and cognitive decline. However, research over the past 30 years has failed to significantly advance our understanding of how tau causes dementia, limiting the design of rational therapeutics. It has become evident that we need to expand our understanding of tau in physiology, in order to delineate how tau may contribute to pathology. This review discusses recent evidence that has uncovered a novel aspect of tau function, based on its previously uncharacterized localization to the synapse. Here, multiple streams of evidence support a critical role for synaptic tau in the regulation of synapse physiology. In particular, long-term depression, a form of synaptic weakening, is dependent on the presence of tau in hippocampal neurons. The regulation of tau by specific phosphorylation events downstream of GSK-3β activation appears to be integral to this signaling role. We also describe how the regulation of synapse physiology by tau and its phosphorylation may inform our understanding of tauopathies and comorbid diseases. This work should provide a platform for future tau biology research in addition to therapeutic design.
Keywords
Introduction
Following the identification of the microtubule-associated protein (MAP) tau as a major component of neurofibrillary tangles (NFTs) in Alzheimer’s disease (AD) brains (Kosik and others 1986; Grundke-Iqbal and others 1986), numerous neurodegenerative disorders were soon classified as “tauopathies,” with tau as a seemingly integral component of neuronal pathology. Such diseases include frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), progressive supranuclear palsy (PSP), Pick’s disease (PiD), corticobasal degeneration (CBD), among many others (Williams 2006). The extent of tauopathy in AD appears to correlate strongly with the degree of cognitive and neuronal decline (Nelson and others 2012), implicating tau as a causative factor for disease progression. Furthermore, mutations and polymorphisms to the tau gene (MAPT) are associated with approximately 5% of frontotemporal dementia cases and are established risk factors for PSP and CBD (Goedert and others 2012; Hutton and others 1998), suggesting that dysregulation of tau itself can indeed cause neurodegeneration. This notion is supported by experimental models of AD, in which the presence of tau is required, and can even be sufficient, for the manifestation of neurotoxicity and neurodegeneration (Fath and others 2002; Ittner and others 2010; Kim and others 2015; Rapoport and others 2002; Roberson and others 2007; Shipton and others 2011; Zempel and others 2013). Reducing tau or limiting tau phosphorylation or aggregation in these models of disease has thus proved to be a highly effective method for reducing the pathological phenotype (Kim and others 2015; Mocanu and others 2008; Noble and others 2005; Serenó and others 2009; Sydow and others 2011; Van der Jeugd and others 2012; Zempel and others 2013). Consequently, an interest in developing tau-based therapeutics for AD patients has been awakened, with a particular focus on tau aggregation inhibitors, tau kinase inhibitors, and microtubule-stabilizing drugs. However, with these current approaches having shown poor or modest efficacy in clinical and preclinical trials, and alternative immunotherapy approaches still in their developmental infancy, a truly effective tau-based therapy seems like a distant reality (Anand and Sabbagh 2015; Mably and others 2015; Pedersen and Sigurdsson 2015).
It seems that despite the conspicuous link between tau and neurodegeneration, our understanding of the pathogenic effects of tau has failed to significantly evolve over recent years and this could ultimately limit the design of rational therapeutics. Overcoming this issue clearly requires an advanced understanding beyond existing hypotheses of how tau affects neuronal biology. It has been suggested that disturbances to the physiological functions of tau could be integral to the triggering of disease processes (Morris and others 2011). Therefore, our current lack of understanding of the relevance of tau to these disease phenotypes likely stems from the shadow of an existing dogma regarding the precise function of tau at a physiological level. Until recently, our knowledge in this regard has remained mostly limited to the role of tau in the establishment of neuronal polarity and axonal elongation, via its microtubule stabilizing capacity (Cáceres and Kosik 1990; Esmaeli-Azad and others 1994). In contrast, very little has been elucidated with regards to the role of tau in mature neurons, aside from its influence over the axonal transport of protein cargoes through effects on microtubule-based motor proteins (Dixit and others 2008). However, failures to observe overt neuronal and behavioral phenotypes in tau knockout mice cast doubt over the absolute physiological significance of these tau roles to neuronal and brain function (Ke and others 2012; Morris and others 2011). With all of this in mind, it is now important to consider recent findings that have begun to uncover a novel role for tau in neuronal physiology, revealing a previously uncharacterized synaptic localization for tau where it acts to regulate synaptic function and plasticity. This review will highlight the evidence collected so far in this regard, and will discuss its implications for brain function and how this could contribute to further understanding of pathological processes and therapeutic advances in tauopathies.
Axonal versus Dendritic Tau: All about Location?
The canonical understanding of tau is that of an axonally segregated protein, whereby it acts to control neurite outgrowth and maintain neuronal polarity, primarily through its effects on microtubule dynamics (Binder and others 1985; Cáceres and Kosik 1990; Hirokawa and others 1988). It had generally been considered to be absent from dendrites apart from in developing neurons (Kosik and Finch 1987) or under pathological conditions (Kowall and Kosik 1987). However, a number of new observations are beginning to dispel this ideology and have led to the premise that tau is also located in dendrites as well as in pre- and post-synaptic structures of healthy neurons. As can be seen in Table 1, overwhelming evidence via numerous methodological approaches now supports this premise.
Evidence for Dendritic and Synaptic Tau in Healthy Neurons.

Abbreviations: cTau = cleaved tau; pTau = phosphorylated tau; hTau = human tau; FRAP = fluorescence recovery after photobleaching; GC = glucocorticoid; GFP = green fluorescent protein; PSD = post-synaptic density; TG = transgenic; WT = wild-type.
It is not entirely clear why the dendritic localization of tau has only recently come to be appreciated. Epitope masking by different phosphorylation tendencies or protein–protein interactions of tau seems to have limited previous analysis of tau distribution (Binder and others 1985). Indeed, the same antibody that failed to detect dendritic tau under normal conditions, readily detects tau in the somatodendritic domain following the removal of tau phosphorylation (Papasozomenos and Binder 1987). This finding, and others (Tashiro and others 1997), suggests that tau proteins exhibit subcellular compartmentalization based on phosphorylation profile, possibly a result of a presence of a cellular gradient of tau kinase activity (Mandell and Banker 1996). It has now also been noted that different tau isoforms (see Box 1) display distinct subcellular localization patterns, with 1N and 2N, but not 0N isoforms present in the dendrites, and an absence of 1N tau in axons (Liu and Götz 2013). This evidence further supports the case for a differential distribution of tau-isoform populations between the axonal and dendritic compartments, in terms of both structure and phosphorylation, which may ultimately pertain to distinct functional roles.
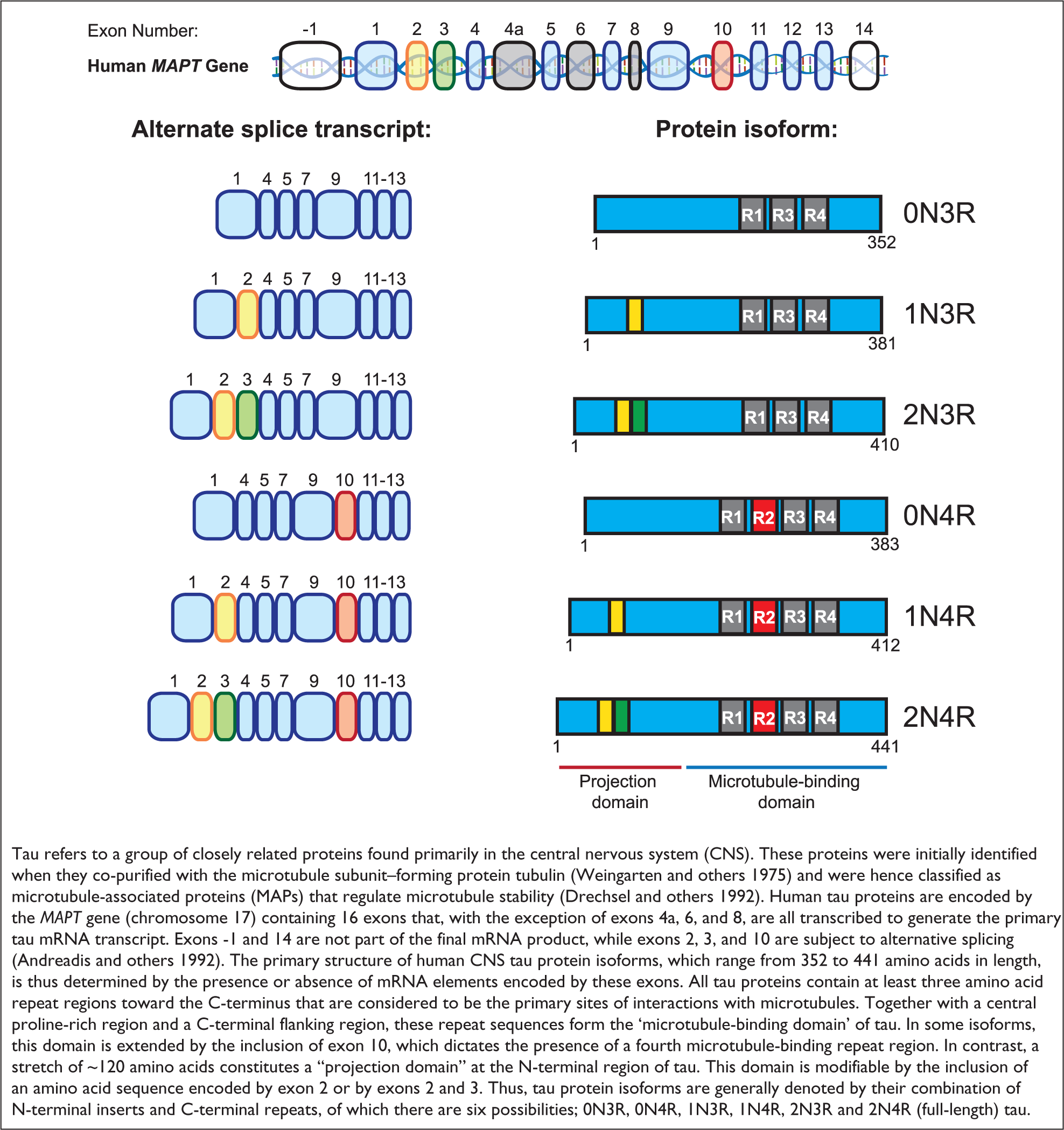
Tau Protein Isoforms
Observations from control human brains revealed that 70% of post-synaptic structures, which decorate dendritic branches, immunostained positive for tau (Tai and others 2012). This makes it all the more remarkable that tau can only recently be said to have become generally accepted as a post-synaptic molecule (see Table 1). A possible explanation for this apparent delayed characterization of tau as a post-synaptic protein may be its potentially short-lived presence within these dendritic microdomains. Indeed, some evidence suggests that tau enters dendritic spines only transiently and in a regulated manner (Frandemiche and others 2014). Such a transient and regulated presence may reflect fleeting microtubule invasion events carrying tau into and out of spines (Hu and others 2008; Jaworski and others 2009), although this possibility has yet to be proven or disproven.
Regardless, the presence of tau in dendritic and synaptic compartments opens up a new and compelling chapter in tau biology, one that is laden with questions that must now be answered. For instance, Mandelkow and colleagues have previously provided evidence for the existence of a diffusion barrier that prevents tau from entering the somatodendritic compartment of mature neurons (Li and others 2011). If true, this begs the question as to how tau can come to be located in these distinct cellular domains. This diffusion barrier is reported to be ineffective at sorting phosphorylated tau, or tau detached from microtubules (Li and others 2011), lending weight once again to the notion that tau in the somatodendritic domain may have a distinct phosphorylation and functional profile to that of axonal tau. The observation that tau mRNA is also found in dendrites and dendritic spines (Malmqvist and others 2014) opens up the possibility that local translation could provide the source of dendritic tau, mitigating the effects of any axonal barrier to protein diffusion. Alternatively, local trans-synaptic uptake of tau protein from the extracellular space, following its activity dependent release from pre-synaptic terminals remains a viable possibility (Pooler and others 2013; Sokolow and others 2015; Yamada and others 2014). Such trans-synaptic spreading of tau could thus be hijacked in disease states, facilitating the propagation of tau pathology between interconnected brain regions (Clavaguera and others 2009; de Calignon and others 2012; Krüger and Mandelkow 2015; Liu and others 2012).
Despite these unresolved issues, the true questions posed by this expanding pool of evidence for dendritic and post-synaptic tau relate to the physiological and functional significance of these findings.
Tau Regulation of Synaptic Function
A number of circumstantial findings indicate that tau itself can regulate synaptic function. For example, tau can directly or indirectly modulate the signaling of synaptic receptors such as muscarinic acetylcholine receptors (mAChRs) and N-methyl-
In order to delineate a possible function of tau at the synapse, Kimura and colleagues studied neuronal function in the hippocampus of tau knockout mice. This brain region has been extensively used to study synaptic plasticity events, such as long-term potentiation (LTP) and long-term depression (LTD) at CA3-CA1 synapses in particular (Andersen and others 2007). In vivo and ex vivo electrophysiological recordings revealed a selective deficit in LTD, but not LTP, from the brains of mice with a homozygous or heterozygous tau deletion, but not from their wild-type counterparts (Kimura and others 2014). Corroborating these findings, acute suppression of tau with tau-shRNA prevented the expression of LTD, and not LTP, in neurons from organotypic cultured hippocampal slices (Kimura and others 2014; Regan and others 2015). An important aspect of this in vitro study was the acute and sparse ablation of tau, largely restricted to CA1 neurons of the hippocampal slice. These factors eliminated any potential confounding developmental effects associated with tau knockout mice and ensured that LTD deficits could not be attributed to alterations of pre-synaptic afferents from the CA3 region, thus implicating post-synaptic tau as the locus for LTD signaling. Replacing the silenced endogenous rat tau with human tau effectively restored the ability of these neurons to exhibit LTD, clearly demonstrating a specific requirement for tau protein in this form of synaptic plasticity (Kimura and others 2014). The absence of any observable LTP deficit is consistent with one study that has shown normal LTP in tau knockout mice (Shipton and others 2011) but contrasted with recent findings from another research group, who showed normal LTD and impaired LTP in the hippocampus of tau knockout mice (Ahmed and others 2014). Furthermore, a recent study showed that LTD was readily inducible across mossy fiber (mf-LTD) synapses in tau knockout mice (Decker and others 2015), raising the interesting question as to how to explain these opposing results. The answer likely stems from features of the different experimental models in terms of the age of the hippocampal slices and the induction protocols used for plasticity (Ahmed and others 2011; Kemp and others 2000), and/or reflects a specialization of tau function at specific hippocampal synapse circuits depending on the locus or mechanisms of synaptic plasticity (e.g., tau-independent mf-LTD is expressed pre-synaptically (Kobayashi and others 1996) whereas tau-dependent LTD at CA3-CA1 synapses is generally regarded as post-synaptic (Collingridge and others 2010)). Therefore, it is undoubtedly important to probe further into a possible age-dependent and synapse-specific involvement of tau in mechanisms of synaptic plasticity induction, which could also provide further understanding into why certain forms of tau lead to neurotoxicity in the matured and/or aged brain.
Together, these studies have unveiled a novel aspect to tau function, one that pertains to the regulation of synapses and their plasticity. Given the explicitly demonstrated role of synaptic plasticity processes in memory and recall (Nabavi and others 2014) it is no surprise that behavioral studies in tau knockout mice show that this deficit in hippocampal LTD correlates with impaired memory performance, in particular hippocampus-dependent spatial reversal learning (Regan and others 2015) (see Box 2). Thus, unlike previous functions attributed to tau, its role in synaptic regulation does not appear to be redundant or functionally compensated in tau knockout mice, underscoring the physiological significance of synaptic tau.
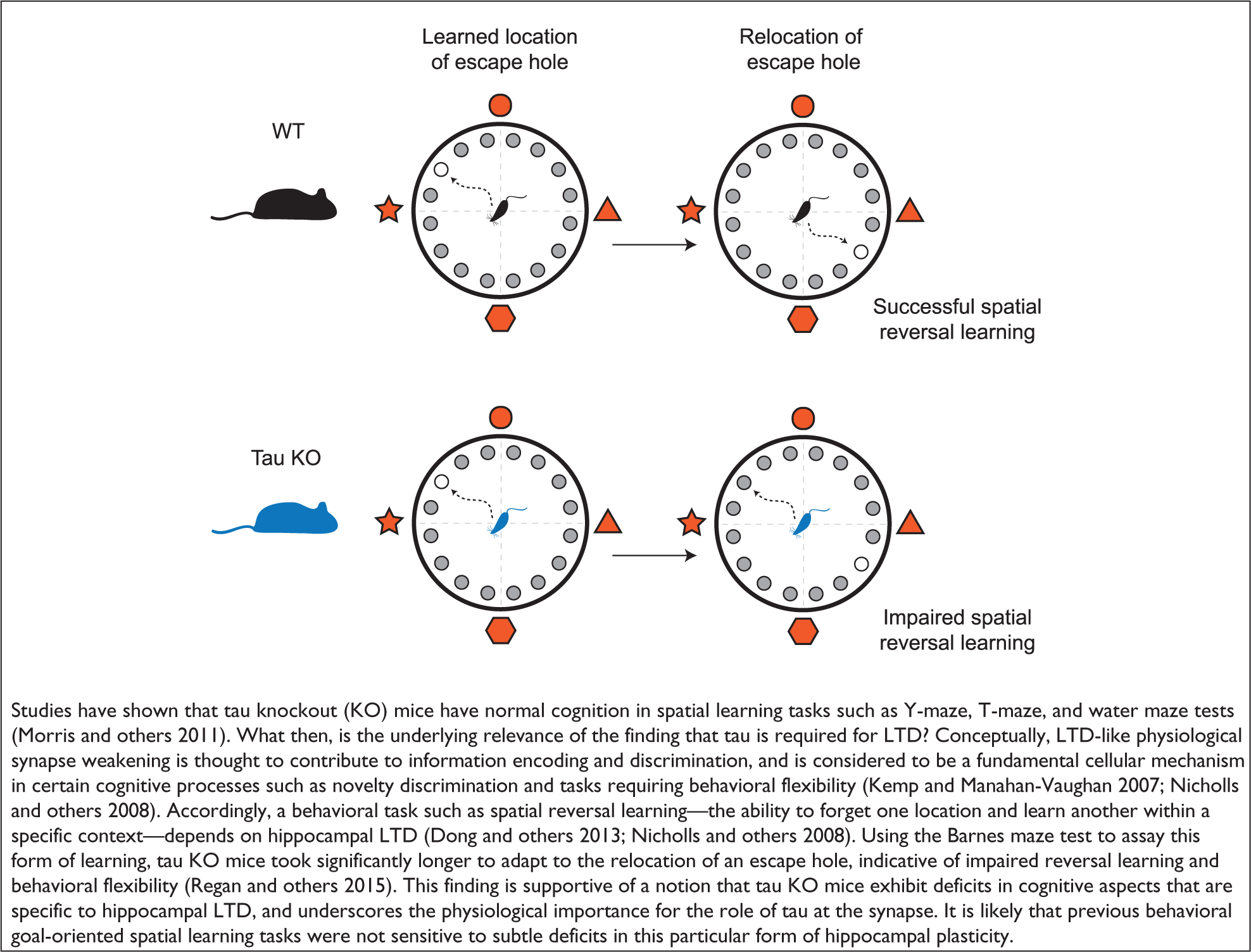
Tau and Cognition
A major remaining challenge is to resolve the capacity in which tau is involved in synaptic plasticity mechanisms within the post-synaptic domain. Our recent study showed that an interaction between protein interacting with C kinase 1 (PICK1) and the GluA2 subunit of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) is impaired during LTD induction in tau knockout mice (Regan and others 2015). Given the importance of this protein interaction for activity-driven AMPAR endocytosis and hippocampal LTD (Citri and others 2010; Hanley and Henley 2005), this is likely to reflect a mechanistic readout of the impaired LTD in tau knockout mice. Recent work using a transfected cell line has now shown that the presence of tau significantly enhances the GluA2-PICK1 interaction, supporting an important role for tau in regulating the association between these two proteins (Yagishita and others 2015). However, the way in which tau might regulate this AMPAR endocytosis mechanism is not known and requires further investigation. Furthermore, tau can also bind to variety of other proteins that are known to be involved in the regulation of synaptic AMPARs (Reynolds and others 2008; Liu and others 2012), making it clear that tau has the potential to affect a number of different signal transduction cascades that pertain to the trafficking of AMPARs and thus the regulation of synaptic function.
The Phosphorylation Signature of tau
Alternative splicing and an array of post-translational modifications (PTMs) converge to make tau an incredibly heterogeneous species of protein (Larcher and others 1992; Morris and others 2015). Phosphorylation is undoubtedly the best-studied form of tau PTM, primarily a result of its indubitable links with tau aggregation and AD pathology (Goedert and others 1992; Morishima-Kawashima and others 1995). However, tau is also highly phosphorylated in the absence of neurotoxicity, such as in the fetal brain, during hibernation, and to a lesser extent in the adult brain (Arendt and others 2003; Matsuo and others 1994; Yu and others 2009). Tau phosphorylation is therefore not simply a pathological event, but clearly serves to regulate tau in a physiological sense. Little has been elucidated in this regard however; phosphorylation is known to affect the interaction of tau with microtubules and other binding partners (Biernat and others 1993; Reynolds and others 2008; Usardi and others 2011), but the actual physiological consequences of these effects are not evident.
As alluded to above, tau species within distinct subcellular domains appear to be characterized by distinct “signatures” of protein phosphorylation (Mandell and Banker 1996; Tashiro and others 1997). Indeed, a recent study has shown that tau within the post-synaptic density is most prominently doubly phosphorylated between resides 386–404 (Morris and others 2015). But how can we relate these distinct phosphorylation signatures to the functional output of tau? Given the extremely labile nature of tau phosphorylation, this question remains fundamentally difficult to answer, but a number of recent insights are beginning to show clear phosphorylation-function relationships for tau, specifically at the synapse.
Much of this knowledge has been ascertained through biochemical analysis of tau with phosphorylation residue-specific antibodies, followed by the analysis of mutant tau proteins with mutations preventing phosphorylation at those specific residues. For example, Frandemiche and colleagues found that chemically induced synaptic activity in cultured neurons resulted in increased tau phosphorylation at threonine 205, and this was associated with the translocation of tau from the dendrite to the synapse. Expressing a mutant form of tau, unable to be phosphorylated at threonine 205, appeared to compromise the stability of synaptically translocated tau following the same synaptic activation paradigm (Frandemiche and others 2014). Intriguingly, the same study found a similar synaptic translocation was induced by treatment of the neurons with amyloid-β, but this was associated with the phosphorylation of tau at serine 404. Thus, although both phosphorylation events appear to relate to synaptic translocation, the signature of tau phosphorylation seems to differ depending on the stimulus.
Our group have shown that electrically driven synaptic activity elicits an increase in tau phosphorylation at serine residues 396 and 404, with no change in phosphorylation at serine 202 and threonine 205 residues (Kimura and others 2014; Regan and others 2015). This finding differs somewhat from the previously described study. While differences in the experimental substrate could be responsible, for example, cultured neurons versus hippocampal slices, an intriguing possibility is that differing forms of synaptic activity evoke differing signatures of tau phosphorylation. Specifically, threonine 205 phosphorylation was evoked by synaptic activity that induces LTP-like synaptic changes (Frandemiche and others 2014), whereas serine 396 and 404 phosphorylation was evoked by an LTD-inducing stimulus (Kimura and others 2014; Regan and others 2015). Inhibitors of NMDARs and GSK-3β blocked this specific change in tau phosphorylation, and prevented the interaction of PICK1 with GluA2. Given the importance of NMDARs and GSK-3β to hippocampal LTD (Dudek and Bear 1992; Mulkey and Malenka 1992; Peineau and others 2007; Peineau and others 2009), these findings affirm the relevance of tau phosphorylation to LTD, and suggest that tau is probably a substrate for GSK-3β in an LTD signaling cascade downstream of NMDAR activation. The expression of mutant forms of tau demonstrated that tau phosphorylation, specifically at serine 396, is fundamentally required for LTD and has no effect on LTP (Regan and others 2015). This finding was unique in highlighting a distinct phosphorylation-to-function relationship at just a single residue of tau. Therefore, it appears that phosphorylation of tau at serine 396 is an integral event in a GSK-3β signaling pathway that mediates synaptic weakening (Fig. 1).
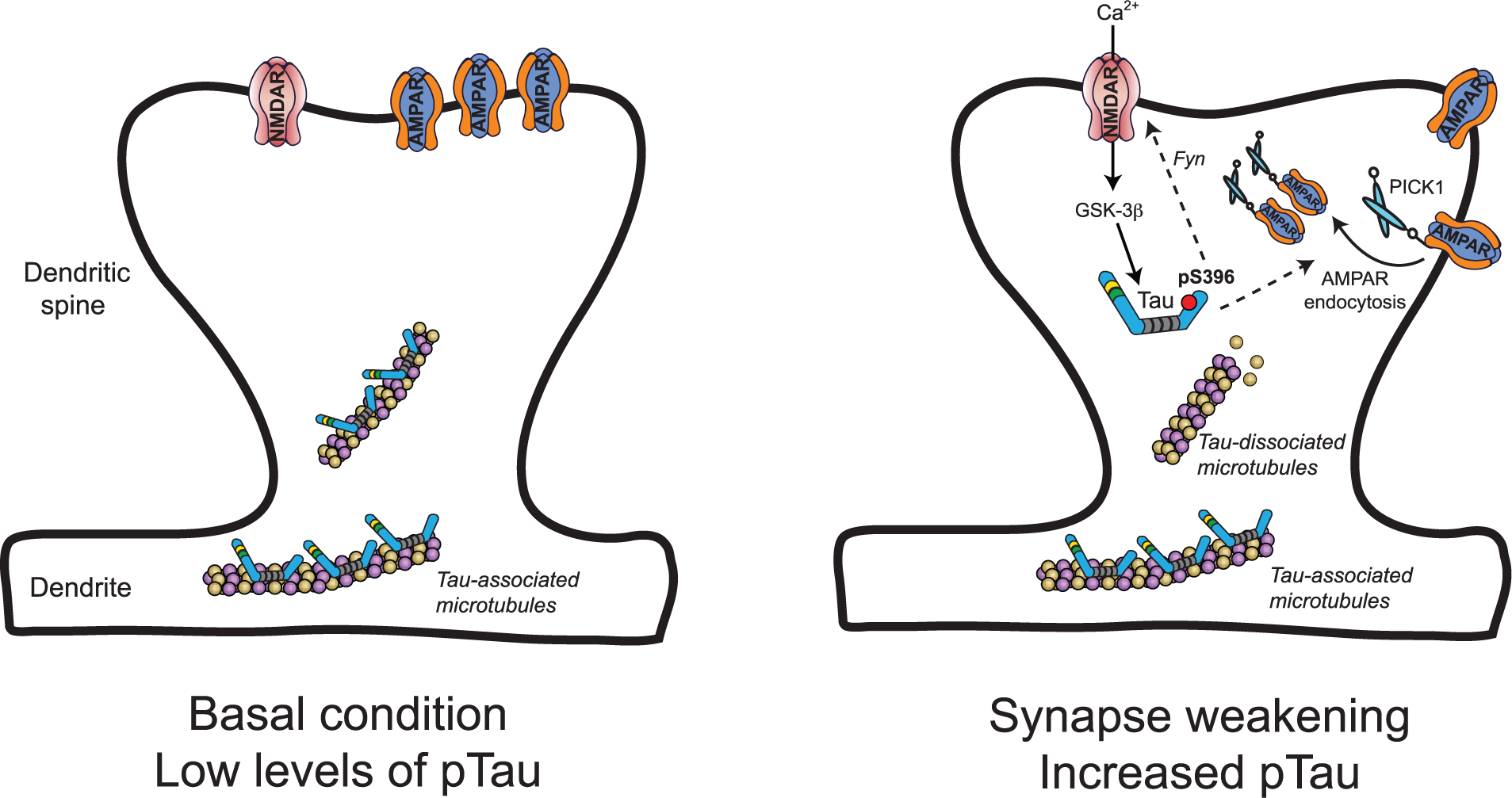
Putative model for the role of tau and tau phosphorylation during synaptic weakening. During basal conditions (left) tau is present in dendrites and dendritic spines, where it is most likely bound to microtubules and has low levels of phosphorylation (pTau). A synaptic weakening stimulus (right) triggers Ca2+ influx through NMDARs that activates signaling cascades involving the serine/threonine kinase GSK-3β. GSK-3β mediates the transient phosphorylation of tau at serine 396, which is a critical event for subsequent AMPAR internalization. Downstream effects of tau phosphorylation depend on its specific interacting partners at the synapse, but may involve changes to microtubule stability and/or the regulation of NMDARs via Fyn trafficking, ultimately regulating the association of an AMPAR-PICK1 complex. AMPAR = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors; GSK-3β = glycogen synthase kinase–3β; NMDAR = N-methyl-
Together, these findings demand further investigation into other phosphorylation signatures associated with and required for synaptic function. Similarly, the mechanisms by which tau phosphorylation can regulate synaptic function ought to be examined. One study suggests that phosphorylation of tau reduces the association of tau and Fyn with the post-synaptic density protein PSD-95 and the NMDAR subunit GluN2B (Mondragón-Rodríguez and others 2012). This supports a previous study informing that tau regulates the association of Fyn with NMDARs (Ittner and others 2010), and suggests that tau phosphorylation may thus modulate NMDAR signaling via reduced GluN2B phosphorylation by Fyn. Data are presented to suggest that phosphorylation of tau at threonine 231 might be a critical regulator of this post-synaptic protein interaction (Mondragón-Rodríguez and others 2012). Marrying this finding with the other observations of synaptically evoked tau phosphorylation signatures will be required to generate the complete picture of the physiological relationship between tau phosphorylation and function at the synapse.
Finally, it will be important to consider the relevance of other tau PTMs when considering the synaptic function of tau. A recent article described the array of other physiological PTMs—including methylation, acetylation, ubiquitination, and O-GlcNAc modification—that are found at tau residues in brain tissue from wild-type mice (Morris and others 2015). These modifications have been relatively poorly studied thus far, but are known to affect microtubule binding, tau aggregation and degradation among other things, as well as cross-talk with other PTMs (Morris and others 2015). Additionally, little is known about the physiological effects of C-terminal cleavage of tau by caspase, which occurs following the activation of GSK-3β and site-specific tau phosphorylation events (Cho and Johnson 2004; Ding and others 2006; Mondragón-Rodríguez and others 2008) and is associated with synaptic dysfunction and memory impairment (Kim and others 2015; Zhao and others 2015). This facet of tau regulation is particularly intriguing given the roles of both caspase and GSK-3β in LTD signaling (Li and others 2010; Peineau and others 2009). Resolving the relationship between these PTMs and tau function remains a clear and fundamental challenge to enhancing our understanding of tau pathophysiology.
A Pathological Perspective for Synaptic Tau
Tau pathology, which is evident in tauopathies such as AD, is generally characterized by three main hallmarks: (a) tau becomes hyperphosphorylated, with the levels of tau-associated phosphate roughly tripling in AD brains compared with healthy adult brains (Köpke and others 1993), (b) tau is re-distributed to the somatodendritic regions of neurons (Grundke-Iqbal and others 1986; Kowall and Kosik 1987), and (c) hyperphosphorylated and cleaved tau proteins fold into structures that form paired-helical filaments, which then aggregate to form insoluble intra-neuronal NFTs (Goedert and others 1988; Grundke-Iqbal and others 1986; Kosik and others 1986). It is generally considered that (a) leads to (b) and (c) to bring about tau pathology. Although amyloid pathology precedes this tau pathology in patients with AD, correlative studies have shown that it is the extent of tau pathology that closely mirrors, and appears to dictate the extent of, neuronal loss and cognitive decline (Murray and others 2015; Nelson and others 2012). However, it has now become evident that NFTs themselves are actually dissociated from the decline in neuronal and cognitive function, and are neither necessary nor sufficient for neurotoxicity (Guerrero-Muñoz and others 2015), arguing against their use as therapeutic targets. In light of this, it is now necessary to elucidate precisely how the hyperphosphorylation of tau can evoke neurotoxicity or synaptic dysfunction.
A long-standing debate concerns whether neurotoxicity is triggered by the generation of a pathological species of tau protein, or whether physiological functions of tau are somehow disrupted; a gain of function or a loss of function. Two recent studies fall either side of this debate: Song and colleagues show that rTg4510 AD-model mice accumulate pathological species of tau carrying multiple PTMs at an early stage of disease progression (Song and others 2015). In contrast, Morris and colleagues provide evidence that tau from hAPP AD-model mice has similar PTMs to physiological tau found in wild-type mice (Morris and others 2015). Clearly the difference in AD models must be playing a significant role in this discrepancy; whilst the rTg4510 model mice overexpress P301L mutant forms of human tau, the hAPP mice overexpress mutant forms of hAPP but with no genetic modifications to tau. Thus, the hAPP mice arguably provide a better model of sporadic tauopathies, that is, the majority of cases of AD, where no mutations to the MAPT gene are present and tau is simply regulated or dysregulated by external stimuli (i.e., as a result of elevated amyloid-β). If we are to consider this hAPP model reliable, therefore, then a reasonable conclusion from the latter study is that a pathological species of tau is unlikely to be responsible for triggering neuropathology in AD, suggesting that dysregulation of the physiological functions of tau is more relevant to disease onset (Morris and others 2015). Moreover, this study suggests that tau hyperphosphorylation in diseased states should be defined by an increase in the proportion of tau molecules that are phosphorylated at given residues, rather than an increase in the number of phosphorylated residues on each tau molecule. To put it another way, hyperphosphorylated tau, at least in the early stages of disease progression, likely reflects exaggerated physiological modifications to tau proteins, rather than the occurrence of new “unphysiological” modifications.
What could this mean for tau function? Given that AD is widely considered to be a synaptic disorder (Scheff and others 2006; Selkoe 2002) and that the synaptic deficits induced by amyloid-β critically depend on the presence of tau (Ittner and others 2010; Rapoport and others 2002; Roberson and others 2007; Shipton and others 2011; Zempel and others 2013), a new perspective for the causality of neurodegeneration by tau can be gained from what is now known about the actual physiological function of tau at the synapse. For example, up-regulation of LTD signaling is considered to be central to aberrant synaptic elimination in AD (Jo and others 2011; Li and others 2010; Regan and others 2014) and amyloid-β has been shown to facilitate LTD (Cheng and others 2009; Hsieh and others 2006; Kim and others 2001; Li and others 2009; Shankar and others 2008). Now we know that tau is intrinsic to LTD, these findings raise the intriguing possibility that synaptic tau function is aberrantly regulated by amyloid-β to enhance LTD signaling. Indeed, amyloid-β induces changes to tau phosphorylation and synaptic translocation that are similar to that induced by synaptic activation paradigms (Frandemiche and others 2014; Mondragón-Rodríguez and others 2012), suggesting that physiological synaptic functions of tau are somehow “hijacked” by amyloid-β. Furthermore, synaptic internalization of AMPARs, a classical LTD expression mechanism, is induced by amyloid-β in a manner that depends on tau phosphorylation (Miller and others 2014). Therefore, one can predict that the aberrant phosphorylation of tau following the amyloid-β driven activation of kinases such as GSK-3β (Lei and others 2011), cyclin-dependent kinase 5 (Cdk5) (Zempel and others 2010), MAPK (Ferreira and others 1997) or AMP-activated protein kinase (AMPK) (Mairet-Coello and others 2013) will lead to the aberrant upregulation of LTD signaling and synaptic weakening in AD. Of these, the principal tau kinase GSK-3β may prove to be especially pertinent, given that its involvement in a critical LTD signaling cascade is known to be facilitated by amyloid-β (Jo and others 2011; Li and others 2010). Here, caspase-3 activation leads to the cleavage of Akt1 and removal of constitutive GSK-3β inhibition. A wealth of evidence supports that amyloid-β and associated neurotoxic insults, such as reactive oxygen species, can activate neuronal caspase-3 (Garwood and others 2011; Marín and others 2000; Narayan and others 2014), indicating that this signaling pathway culminating in GSK-3β-mediated phosphorylation of tau could be integral to the shift toward pathological synaptic weakening and synapse loss (Fig. 2).
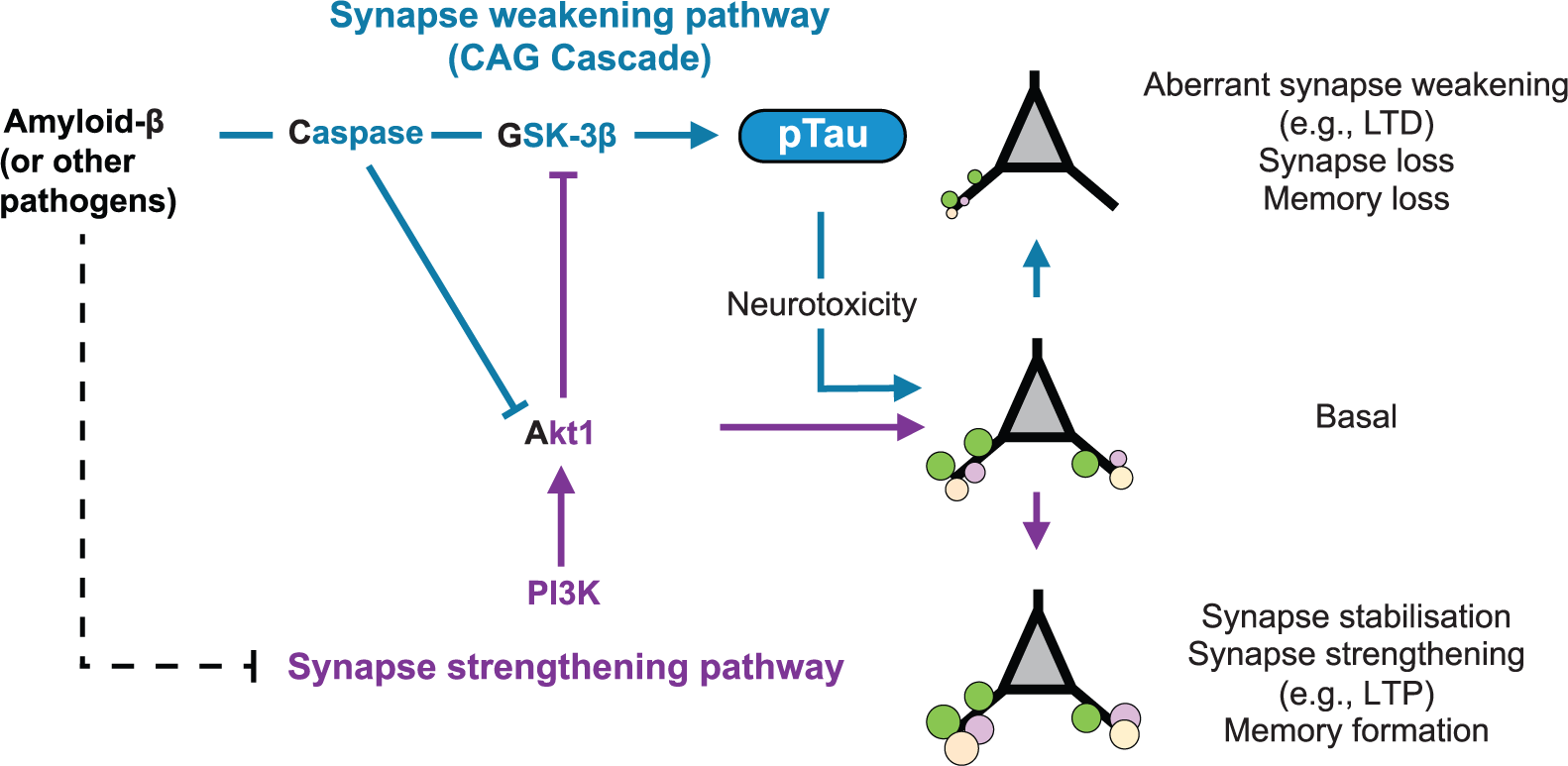
Conceptual schematic illustrating synapse pathology in tauopathies. Neurotoxic pathogens such as amyloid-β aberrantly activate a synapse weakening CAG signaling cascade and simultaneously inhibit synapse strengthening pathways, together converging on the hyperphosphorylation of tau (pTau). pTau at the synapse drives excessive long-term depression (LTD) signaling, resulting in a shift from the basal state toward pathological synapse loss and neurodegeneration.
In this context, it is particularly interesting to note that phosphorylation of tau at serine 396, which we now know to be critical for LTD (Regan and others 2015), has been shown to be an early event in AD pathogenesis (Mondragón-Rodríguez and others 2014) and that tau phosphorylated at serine 396 has been shown to accumulate in the post-synaptic compartments of AD brains and other tauopathies at an early stage (Muntané and others 2008; Tai et al., 2012). Once again, these findings suggest that early synaptic loss in neurodegenerative diseases such as AD could be mediated through the aberrant enhancement of a physiological tau phosphorylation signal at the synapse.
Dementia Comorbidities: Tau as the Missing Link?
On a final note, it is worth considering evidence that tau hyperphosphorylation is a common response to many neuronal stressors, such as hypothermia, glucose deprivation (Planel and others 2004; Yanagisawa and others 1999), diabetes (Platt and others 2015; Wang and others 2014), and chronic stress (Sotiropoulos and others 2011). These findings highlight an intriguing molecular crossover between neurodegenerative pathways and the effects of other changes to the biological environment that are considered to precipitate the onset of dementia. For example, chronic stress and diabetes are thought to contribute to a neurodegenerative phenotype, as evidence by their considerable comorbidity in both patients and experimental models (Artola 2008; Janson and others 2004; Magariños and McEwen 2000; Srivareerat and others 2009; Stranahan and others 2008; Verdile and others 2015). In particular, experimental models have shown that chronic exposure to stress hormone glucocorticoids facilitates LTD signaling (Kim and others 1996; Xu and others 1997; Yang and others 2004) and induces tau accumulation at the synapse, with a specific increase in serine-396-phosphorylated tau within the synaptic compartment (Pinheiro and others 2015). Furthermore, a similar effect has been shown with the proBDNF valine 66 (Val66) isoform, which conveys an increased susceptibility to cognitive decline and AD in late life, as compared to its methionine 66 (Met66) isoform (Harris and others 2006; Ventriglia and others 2002; Voineskos and others 2011). Treatment of brain slices with proBDNF Val66 facilitates LTD and this is mediated via the activation of GSK-3β and associated with an increase in tau phosphorylation at serine 396/404 (Kailainathan and others 2016). Together, these findings indicate a convergence of external synapse weakening/death factors on synaptic tau function that could potentially “prime” a diseased brain state and promote the onset of dementia.
Conclusion
Recent work revealing the function and regulation of tau at the synapse has strayed beyond the existing dogmatic approach to tau research. Tau function is clearly far more diverse than has largely been considered, with tau taking on multiple roles within the neuron. It will be essential to incorporate this new level of understanding into designing rationale for therapeutic approaches aimed at tau, if we are to facilitate the development of efficacious treatments for tauopathies.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors’ research was supported by the Medical Research Council (MRC; 984406), the Biotechnology and Biological Sciences Research Council (BBSRC; BB/N001893/1), and the Wolfson Research Merit Award from the Royal Society London.