
Abstract
The understanding of the functional and structural changes occurring in the cerebral cortex and basal ganglia in Huntington’s disease (HD) has benefited considerably from the generation of genetic animal models. Most studies of synaptic alterations in HD models have focused on the striatum, but a more complete picture of synaptic dysfunction in the cortico-basal ganglia-cortical loop is emerging. Here, we provide a review and analysis of current developments in the study of synaptic alterations in these areas using HD rodent models. Recent evidence indicates that cortical maldevelopment plays a role in synaptic dysfunction along the corticostriatal pathway that may have its roots in the way mutant huntingtin interacts with synaptic proteins. Furthermore, a progressive disconnection in the corticostriatal pathway leads to abnormal function engaging extrasynaptic N-methyl-D-aspartate glutamate receptors that contribute to eventual cell degeneration. In addition, biphasic increases followed by decreases in glutamate and dopamine release in the striatum could explain contrasting symptomatology in early and late stages of the disease. Changes in striatal output regions also are beginning to be examined. Finally, we highlight some therapeutic avenues aimed at rescuing synaptic dysfunction.
Introduction
Huntington’s disease (HD) is a heritable neurodegenerative condition caused by a mutation in the Huntingtin (HTT) gene, consisting of an elongation in CAG (glutamine) triplet repeats (Huntington’s Disease Collaborative Research Group 1993). There are excellent reviews characterizing the main symptoms of HD in humans and animal models (Bates and others 2015; Levine and others 2004; Pouladi and others 2013; Ross and Tabrizi 2011). Briefly, the main feature of HD in humans is chorea, which consists of abnormal dance-like movements. Other prominent, disabling features include motor incoordination, impaired balance, dysarthria, and dysphagia (Chan and others 2019; Heemskerk and Roos 2011). Motor symptoms are generally preceded by, or occur simultaneously with, psychiatric disturbances such as depression and anxiety (Duff and others 2007; Paulsen and others 2008), as well as cognitive deficits, in particular frontal dysexecutive syndrome (Ho and others 2003; Lawrence and others 1996). In the late stages of the disease, bradykinesia and rigidity are the predominant motor symptoms. The principal cell type that degenerates in HD is the striatal medium-sized spiny neuron (MSN), also known as striatal projection neuron (SPN). Cortical pyramidal neurons (CPNs) and, to a lesser extent, neurons in other brain regions also are lost (Vonsattel and others 1985; Waldvogel and others 2015).
The introduction of selective neurotoxins facilitated the creation of the first models of HD. Excitotoxins like kainic or quinolinic acid, and mitochondrial toxins, such as 3-nitropropionic acid produced striatal lesions similar to those occurring in HD (Beal and others 1986; Coyle and Schwarcz 1976). Although these models replicated some of the ultimate histopathology of HD, they lacked its progressive course. The first transgenic mouse models, the R6/2 and R6/1 expressing exon 1 of the HD gene, were generated after the HD mutation was identified (Mangiarini and others 1996). Subsequently, a variety of transgenic and knock-in models were developed (Levine and others 1999; Menalled and others 2002; Slow and others 2003; Table 1). Although these models possess construct validity, replication of the human phenotype in rodents has been elusive. One of the limitations of traditional genetic rodent models is that they generally do not exhibit choreic movements (Pouladi and others 2013). This includes the most widely used mouse models, R6/2, YAC128, Q175, and BACHD (Gray and others 2008; Mangiarini and others 1996; Menalled and others 2012; Slow and others 2003), although there are some exceptions (Gu and others 2015) especially in older rodents (Zeef and others 2014). Additionally, some nonhuman primate models of HD may present with dystonia and chorea (Chan and others 2015). These abnormal movements are partially relieved by tetrabenazine (TBZ), which is used to alleviate chorea in HD patients (Huntington Study Group 2006).
Some of the most widely used rodent models of HD and temporal course of motor alterations.

Although most genetic mouse models do not show prominent neurodegeneration or it occurs at the late stage (Turmaine and others 2000), neuronal dysfunction occurs early and underlies disease phenotype. In particular, intrinsic and synaptic neuronal properties are altered when the first overt symptoms emerge (Levine and others 2004). This is not surprising as the mutation is present throughout development. The present review focuses on synaptic dysfunction in the cortex and basal ganglia of HD mice. Due to space and bibliography limitations, we cannot cite all the relevant studies and we apologize for omissions. Similarly, alterations in synaptic plasticity, a natural outcome of synaptic dysfunction, will not be reviewed here and we direct the reader to excellent reviews (Calabresi and others 2016; Plotkin and Surmeier 2015; Smith-Dijak and others 2019).
Huntingtin and Synaptic Function
HTT acts as a scaffold protein involved in vesicular and brain-derived neurotrophic factor (BDNF) transport, cell division, and ciliogenesis (Barnat and others 2017; Saudou and Humbert 2016). HTT also is critical for normal brain development and synaptic transmission (Reiner and others 2003; Smith and others 2005). The evidence for synaptic dysfunction in HD mouse models is overwhelming and pervasive (Chen and others 2018; Joshi and others 2009; Morton and others 2001). So much so that investigators rightly wonder if HD, in reality, is a synaptopathy (Li and others 2003). From a systems perspective this is not entirely surprising. The cortico-basal ganglia-thalamocortical loop is tightly interconnected (Box 1), such that any perturbation at the single-cell level, including nonneuronal elements, likely affects the whole system. HTT is ubiquitous in all brain regions and particularly abundant in the cerebral cortex (layers III and V) and hippocampus. Surprisingly, expression in striatum is not as prominent, suggesting that the levels of expression do not necessarily correlate with neuronal susceptibility to the HD mutation (Reiner and others 2011). In fact, the enrichment of mHTT aggregates in CPNs suggests that the HD mutation may render corticostriatal neurons destructive to their targets more than MSNs being autonomously vulnerable (Fusco and others 1999). This idea agrees with a number of studies providing evidence that the cerebral cortex is necessary for full expression of HD pathology and symptoms (Estrada-Sanchez and others 2015; Wang and others 2014). Thus, identifying the pathophysiological mechanisms in CPNs and how they impact their projection sites, especially in the striatum, is a crucial task (Creus-Muncunill and Ehrlich 2019; Estrada-Sanchez and Rebec 2013).
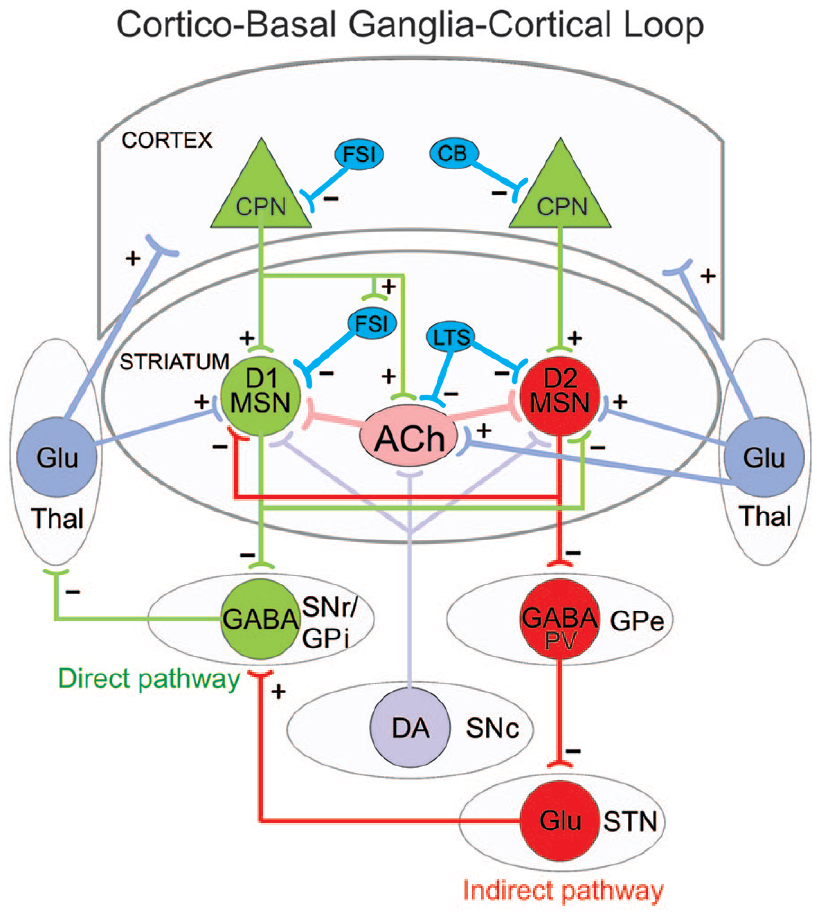
Simplified diagram of the cortico-basal ganglia-thalamocortical loop. Striatal medium-sized spiny neurons (MSNs) expressing dopamine (DA) D1 receptors receive glutamatergic (Glu) inputs (+) from cortical pyramidal neurons (CPNs) and project to the substantia nigra pars reticulata (SNr) as well as the internal segment of the globus pallidus (GPi). This is called the direct pathway and promotes movement. MSNs expressing DA D2 receptors also receive inputs from CPNs and project to the external segment of the globus pallidus (GPe). The GPe, in turn, projects to the subthalamic nucleus (STN) and this to the SNr/GPi. This is known as the indirect pathway and counteracts movement. Both D1 and D2 MSNs also receive afferents from the substantia nigra pars compacta (SNc) and thalamus (Thal). Several classes of interneurons, mainly fast-spiking, parvalbumin (PV)-expressing interneurons (FSI), low-threshold spiking (LTS), and calbindin (CB) GABAergic interneurons exert inhibitory control (−) on CPNs and MSNs. For the sake of simplicity, many synaptic connections are not illustrated. Only those more pertinent for the review are depicted.
However, while most studies of synaptic alterations in animal models have focused, for obvious reasons, on the striatum, a more complete picture of synaptic dysfunction in the cortex and other basal ganglia nuclei is emerging. A comprehensive analysis of these structures will help unravel, for the first time, a long-standing question of whether HD phenotypes develop simultaneously and independently (i.e., cell-autonomously) or as a cascade of heterochronous dysfunction via altered synaptic transmission (Gu and others 2007; Kim and others 2011). The current consensus is that both mechanisms contribute to cell dysfunction and death (Creus-Muncunill and Ehrlich 2019; Lee and others 2013a). The temporal sequence of events leading to the preferential degeneration of particular cell types is more difficult to establish. Based on current knowledge, we will provide evidence and argue that disrupted cell-to-cell communication plays a critical role in HD symptomatology and that correcting synaptic dysfunction is one key for successful therapeutic interventions. We will also contend that, due to the progressive nature of the disease, synaptic transmission may undergo biphasic changes due to compensatory neuroadaptations.
Important Issues
The present review will address a number of issues from an electrophysiological perspective: functional changes leading to corticostriatal alterations and the potential role of early development; glutamate release along the corticostriatal pathway; the role of thalamic inputs to the striatum; alterations in striatal interneurons and internal striatal connectivity; how MSNs of both the direct and indirect pathway affect their targets in the substantia nigra pars reticulata (SNr) and external globus pallidus (GPe), respectively; alterations in the role of dopamine (DA) and neuronal activity in SNr and GPe; and approaches to rescuing synaptic dysfunction.
The Cerebral Cortex
The role of the cerebral cortex and thalamocortical inputs in sensory, cognitive, and motor deficits has, for the most part, been understudied in HD. Of particular importance, there is evidence that many pathological changes in the cortex precede or occur in parallel to those in the striatum (Rosas and others 2003) and subsets of cortical and thalamic neurons are lost in individuals with HD. This suggests that early symptoms may involve altered cortical and, by association, thalamocortical function. The different patterns of cell loss appear to determine the expression of HD symptoms in humans (Waldvogel and others 2015). For example, CPN loss in the primary motor cortex associates with motor dysfunction whereas cell loss in the anterior cingulate cortex relates to mood symptomatology (Thu and others 2010). In addition, morphological and immunohistological studies demonstrated significant interneuron loss, which was region- and symptom-dependent. In the human primary motor cortex there was a selective loss of calbindin-D28k (CB) interneurons in cases with major motor disorder but not in cases with major mood disorder. In contrast, in the anterior cingulate cortex there was a significant loss of CB, calretinin, and parvalbumin-expressing (PV) interneurons in cases with major mood disorder but not in cases with major motor disorder (Kim and others 2014).
In genetic animal models, cortical neuron loss is not as apparent as in human HD. Nevertheless, CPNs undergo morphological alterations such as spine loss and dendritic abnormalities including beading and misaligned apical dendrites (Laforet and others 2001). Functionally, layer II/III neurons in the perirhinal cortex in the R6/1 mouse model display abnormal membrane depolarization and reduced cell capacitance supporting membrane loss and degenerative changes (Cummings and others 2006). Electrophysiological studies in symptomatic R6/2, YAC128 and CAG140 models, demonstrated an increased excitatory drive onto CPNs in layers II/III of the somatosensory cortex (Cummings and others 2009). In contrast, inhibitory drive was drastically reduced, leading to cortical hyperexcitability (Fig. 1). As a result, in R6/2 mice blockade of GABAA receptors (R) induced complex paroxysmal discharges in slices and seizures in vivo. These findings are consistent with interneuron dysfunction and loss in HD. In BACHD mice, although cortical function is intact at 2 months, at 6 months when motor dysfunction has begun there is already reduced inhibition onto CPNs along with reduced excitation onto PV-expressing interneurons suggesting that, in this model, cortical circuitry may develop normally but, with disease progression, synaptic pathology arises (Spampanato and others 2008). However, as discussed in the next section, recent studies indicate that cortical development is not completely normal, at least in some severe HD mouse models (Cepeda and others 2019).
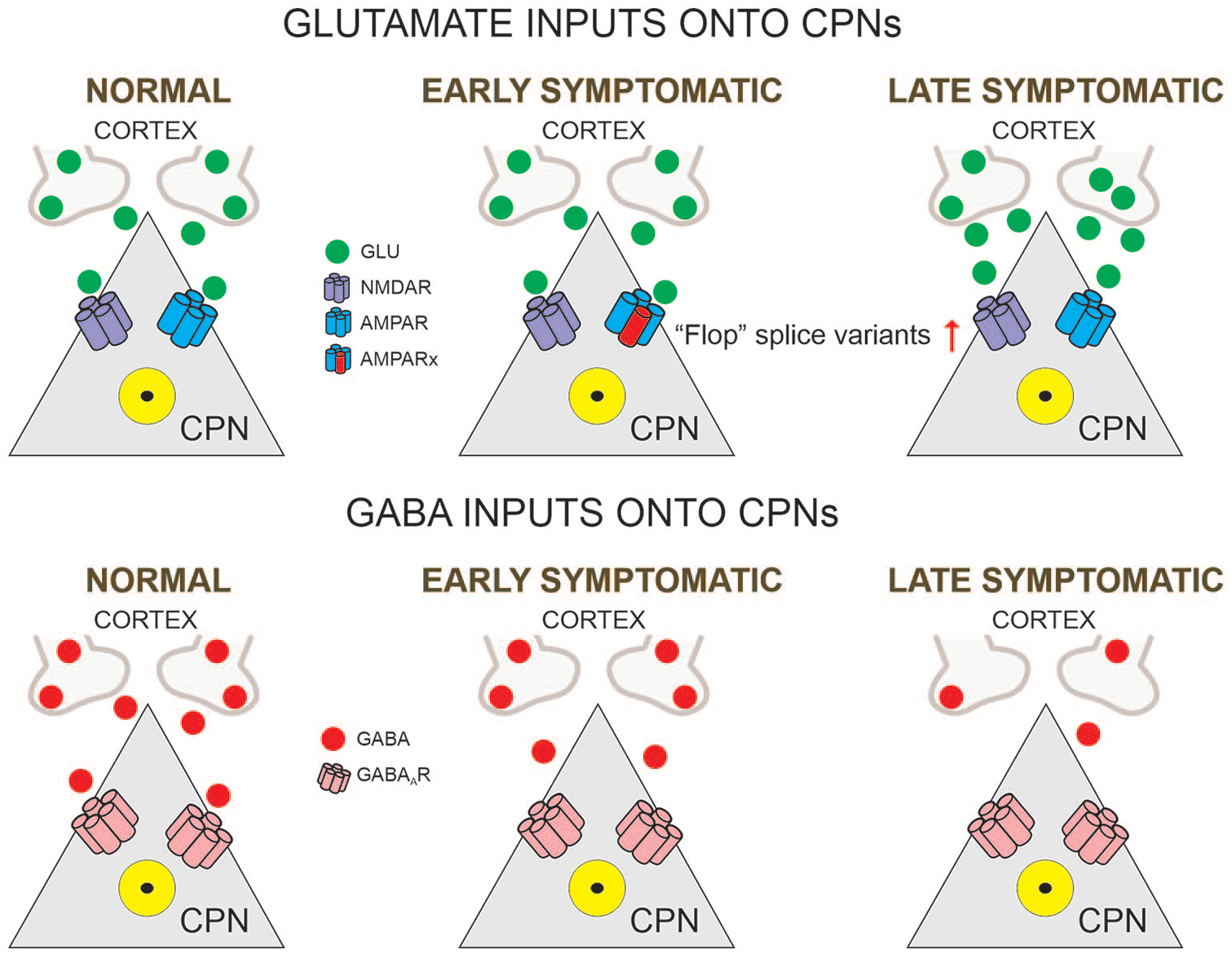
Glutamatergic and GABAergic inputs onto CPNs (cortical pyramidal neurons). In normal conditions, CPNs maintain the excitatory-inhibitory balance. In the early symptomatic stage, glutamate release is not affected but postsynaptic AMPA receptors express the “Flop” variant instead of the “Flip” variant. At this stage, GABA inputs start to decrease. In the late symptomatic stage, glutamate activity shows slight increases and GABA activity is greatly reduced. Alterations in the excitatory-inhibitory balance lead to cortical hyperexcitability. In this and following figures the Rx besides the receptor subtype indicates a modified receptor.
Associated with alterations in biophysical membrane properties and synaptic activity, the firing patterns of CPNs are significantly disrupted in HD models. Extracellular single-unit recordings in prefrontal cortex of freely moving R6/2 transgenic mice demonstrated that spike trains were less variable and had a faster interspike rate than in wildtype (WT) littermates but, at the same time, showed less bursting activity (Walker and others 2008). Remarkably, synchrony between neuronal pairs was significantly attenuated in HD mice, suggesting a population-level alteration of cortical information processing. In support, in vivo Ca2+ imaging of CPN populations in the motor cortex of awake, behaving R6/2 mice revealed that Ca2+ transients (a surrogate of neuronal firing) in symptomatic mice were reduced in amplitude, confirming decreased bursting activity. In addition, correlation coefficients among the occurrence of Ca2+ transients also were decreased, suggesting disrupted neuronal communication in HD cortex (Donzis and others 2019). Interestingly, in presymptomatic Q175 mice, Ca2+ transient frequency was increased. In a similar study, an increase in Ca2+ transient activity occurred abruptly within 1 week before the onset of motor deficits (Burgold and others 2019). Furthermore, the same study demonstrated a pronounced downregulation of synaptic proteins. Of primary importance, histological analyses in R6/2 mice and human HD cases revealed a reduction in perisomatic inhibitory synaptic contacts (Burgold and others 2019). Although a causal link between reduced inhibitory contacts and cortical network dysregulation could not be firmly established in that study, as both appeared simultaneously (around 8 weeks of age in R6/2 mice), it is tempting to speculate that reduced inhibition leads to increased CPN activity. This is supported by in vitro studies demonstrating disturbed excitatory-inhibitory balance as an important pathophysiological mechanism (Cummings and others 2009; Spampanato and others 2008).
Alterations in glutamate receptor-mediated currents also were demonstrated in our studies on dissociated CPNs. α-Amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) and N-methyl-D-aspartate (NMDA) currents were reduced in presymptomatic and early symptomatic R6/2 mice but this difference did not occur in late symptomatic cells (Andre and others 2006). Furthermore, in R6/2 mice more neurons displayed desensitizing AMPA currents, indicating increased expression of “flop” splice variants, unlike WT cells, which expressed “flip” variants. These findings provide additional evidence that cortical dysfunction is an early symptom of HD and that treatment of cognitive deficits could involve AMPAR modulators, such as the ampakines, to selectively increase AMPA currents (O’Neill and others 2004).
Abnormal Cortical Brain Development in HD
There is evidence that the wildtype HTT protein is involved in different aspects of cortical development. For example, in vivo inactivation of HTT alters cell fate of cortical progenitors as well as the polarization and migration of newly generated neurons (Barnat and others 2017; Molina-Calavita and others 2014). HTT loss-of-function during brain development also alters neuronal connectivity. For example, HTT silencing in the developing mouse cortex leads to exuberant cortical synaptic connectivity at 3 weeks of age but deteriorates a few weeks later (McKinstry and others 2014). Furthermore, conditional Emx1-driven deletion of HTT from CPNs reduces neuron numbers in cortex and striatum, without affecting cortical lamination (Dragatsis and others 2018). Similarly, loss of HTT in direct and indirect pathway striatal neurons disrupts synaptic connectivity and recapitulates features of HD, that is, hyperactivity when deleted from indirect pathway MSNs and hypoactivity/motor incoordination when deleted from direct pathway MSNs (Burrus and others 2020). Interestingly, ablation of WT HTT from cells expressing subpallial Nkx2.1 lineage marker, which give rise to PV-expressing interneurons, as well as other types of interneurons, elicits anxiety-like behaviors, hyperkinesia and age-dependent motor deficits (Mehler and others 2019). It is important to indicate that some of the deleterious effects of WT HTT deletion during development are recapitulated by the presence of mHTT. Indeed, expression of mHTT during early development is sufficient to produce a permanent HD phenotype even if expression is terminated at postnatal day 21 (Arteaga-Bracho and others 2016; Molero and others 2016).
During early postnatal brain development in juvenile (R6/2) and adult-onset mouse models of HD, diffuse proto-aggregates have been observed in developing axonal tracts (Osmand and others 2016). These axonal aggregates could alter synaptic physiology. For example, using corticostriatal co-cultures from YAC128 mice, synaptic transmission was impaired as early as 3 weeks in vitro (Buren and others 2016). Recently, we provided evidence that CPNs from R6/2 mice are hyperexcitable and display dysmorphic processes as early as postnatal day 7 (Cepeda and others 2019). In addition, some symptomatic mice present with anatomical abnormalities reminiscent of human focal cortical dysplasia (Blumcke and others 2011), which could explain the occurrence of epileptic seizures in this genetic mouse model and in children with juvenile HD (Cepeda and others 2019; Cummings and others 2009). These studies provide strong evidence that HD is not only a neurodegenerative disorder but also a disease of faulty neurodevelopment (Barnat and others 2020; Blockx and others 2012; Wiatr and others 2018), particularly expressed in the juvenile form of HD (Tereshchenko and others 2015; van der Plas and others 2020).
The Corticostriatal Pathway
If, as recent evidence seems to indicate, cortical development is abnormal in HD, one could expect neuronal alterations and adaptations in cortical target structures, particularly in the striatum. During a critical period of mouse brain development (P10–18), corticostriatal connectivity is extremely sensitive to alterations in cortical activity; thus, early imbalances in cortical function could impair basal ganglia circuit development and function (Peixoto and others 2016). If the excitatory-inhibitory balance in the cerebral cortex of HD mice is disrupted during development, abnormal synaptic activity along the corticostriatal pathway would ensue.
The prevalent hypothesis to explain striatal neurodegeneration posits that excessive release of glutamate onto MSNs and increased sensitivity of NMDA postsynaptic receptors are the main culprits (DiFiglia 1990). Excitotoxicity models of HD could not address this question because these models do not replicate the progression of the disease. After the first genetic models were generated, this question could be explored. Surprisingly, pioneering studies demonstrated a progressive loss of glutamatergic inputs onto MSNs (Cepeda and others 2003; Klapstein and others 2001; Laforet and others 2001; Fig. 2). This loss was accompanied by increases in MSN cell membrane input resistance and reduced capacitance, associated with loss of dendritic spines and reduced somatic areas. The only evidence for increased glutamate transmission was the transient occurrence of large-amplitude synaptic events in presymptomatic and early symptomatic mice (Cepeda and others 2003). These events were hypothesized to reflect increased cortical excitability. In slowly progressive models (e.g., YAC128), changes in glutamate transmission appeared to be biphasic, with early increases followed by decreases (Joshi and others 2009). Early increases were more selective onto D1 MSNs, whereas late decreases affected both types of projection neurons (Andre and others 2011). This demonstrated, first, that synaptic dysfunction in D1 MSNs occurs early in the disease and, second, that there is a progressive, generalized disconnection between cortex and striatum (Cepeda and others 2007), which has been observed in other genetic mouse models as well (Heikkinen and others 2012; Indersmitten and others 2015). Early alterations in excitability and morphology of indirect pathway MSNs also occur in R6/2 and Q175 mice before the onset of motor deficits (Sebastianutto and others 2017). The loss of synaptic contacts could have deleterious consequences on striatal cell survival. For example, there is strong evidence that activation of synaptic NMDA receptors (NMDARs) is neuroprotective, whereas activation of extrasynaptic NMDARs promotes cell death (Hardingham and Bading 2010). This lack of communication (Cepeda and others 2007) or miscommunication (Veldman and Yang 2018) between cortex and striatum, in conjunction with altered postsynaptic responses and NMDAR function imbalance (Raymond and others 2011), could be one of the precursors of neuronal toxicity and the HD phenotype. Another adverse consequence of the progressive corticostriatal disconnection is the loss of important trophic factors, for example, BDNF, critical for MSN support and normal function (Zuccato and Cattaneo 2009).
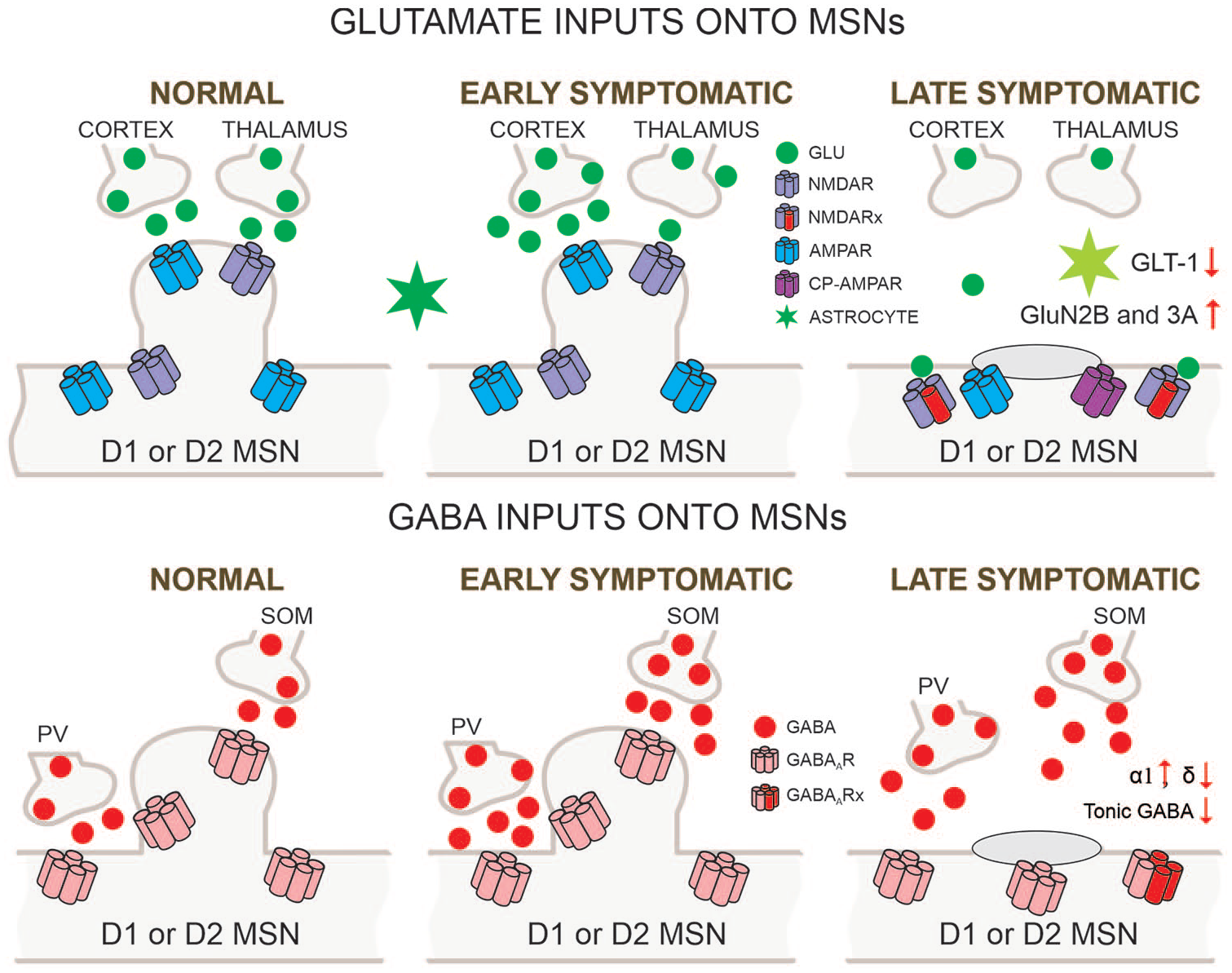
Glutamatergic and GABAergic inputs onto MSNs (medium-sized spiny neurons). Biphasic changes in glutamate release along the corticostriatal pathway occur in HD (Huntington’s disease) models. After initial increases in the early stage, progressive reductions are observed so that in the late stage glutamate inputs from cortex and thalamus are obliterated. In addition, astrocyte function is diminished as demonstrated by reduced glutamate transporter GLT-1. Postsynaptic changes in glutamate receptors also are seen as indicated by altered subunit composition, receptor location, and the presence of Ca2+ permeable (CP)-AMPARs. In contrast, probably as a compensatory mechanism, GABA synaptic activity in MSNs increases. This effect is partly mediated by increased activity of parvalbumin (PV)- and somatostatin (SOM)-expressing GABAergic interneurons. GABAA receptor subunits underlying phasic GABA currents increase whereas those mediating tonic currents decrease.
While MSN degeneration could certainly be facilitated as a consequence of decreased BDNF and activation of extrasynaptic NMDARs, the role of other glutamate receptors in HD such as AMPARs has been examined less frequently. When the GluA2 subunit is present, it renders the channel impermeable to Ca2+, but when GluA2 is absent it becomes Ca2+ permeable (CP), thus named CP-AMPARs. In HD, a decrease in GluA2 subunits has been observed in the putamen from postmortem brain tissue, suggesting alterations in AMPAR-mediated synaptic transmission in this region. CP-AMPARs with a high conductance to Ca2+ could consequently play a predominant role in the motor symptoms of HD (Fourie and others 2014) and in neuronal damage (Mandal and others 2011; Rocher and others 2016). In BACHD mice, for example, the ratio of Gria1/Gria2 mRNA (coding for AMPAR subunits GluA1 and GluA2) is higher than in controls, suggesting the presence of CP-AMPARs (Rocher and others 2016). Furthermore, in human HD patients, the unedited form of Gria2 (indicating high Ca2+ permeability) is higher than in controls (Akbarian and others 1995). Faster decay kinetics, along with reduced amplitudes, of spontaneous AMPAR-mediated currents also have been observed in MSNs from young YAC128 mice compared with controls, suggesting that alternative splicing of AMPAR subunits is different in HD mice, thus leading to the hypothesis that faster decay kinetics could reflect increased flop-containing AMPARs driven by reduced corticostriatal signaling (Milnerwood and Raymond 2007). For example, increased striatal flop-variant AMPAR subunit mRNA expression occurs following cortical deafferentation (Wullner and others 1994). At the circuit level, these changes in subunit expression could reflect compensatory mechanisms resulting from corticostriatal disconnection. Interestingly, increased flop expression and accelerated EPSC decay rates normally occur during development (Koike-Tani and others 2005).
The Thalamostriatal Pathway
Another source of glutamatergic inputs onto the striatum originates in the centromedian-parafascicular (CM/Pf) nuclear complex of the thalamus (Fig. 2). Much less is known about thalamostriatal connectivity than corticostriatal connectivity. Anatomical studies in late-onset mouse models demonstrated early changes along this pathway. In fact, changes in thalamostriatal projection appeared to precede the loss of corticostriatal projections. In the striatum, vesicular glutamate transporter 1 (VGLUT1) expressing terminals emanate from the cortex while VGLUT2 expressing terminals emanate from the thalamus (Fremeau and others 2001). Using VGLUT1 and VGLUT2 immunohistochemistry to detect corticostriatal and thalamostriatal terminals, respectively, a significant reduction in both axodendritic and axospinous thalamostriatal terminals was already evident in the dorsolateral striatum of Q140 mice at 1 month of age and persisted up to 12 months, whereas VGLUT1 terminals appeared normal (Deng and others 2013). Such an early change supports the idea of abnormal development of basal ganglia connectivity (Reiner and Deng 2018). Functional studies using selective optogenetic stimulation of thalamostriatal afferents demonstrated that thalamic afferents to the striatum are affected prior to the overt HD phenotype in YAC128 mice demonstrating that while glutamate release probability was increased, the AMPAR/NMDAR current ratios were decreased (Kolodziejczyk and Raymond 2016). In symptomatic R6/2 mice, we also observed a higher probability of glutamate release at thalamostriatal synapses (Parievsky and others 2017). Furthermore, although peak amplitudes of AMPA- and NMDAR-mediated responses were reduced, consistent with less density of excitatory synaptic contacts, the response areas representing total charge remained unchanged due to an increase in decay times, probably as a compensatory adaptation. Notably, both studies using optogenetic stimulation found increased activation of extrasynaptic NMDARs in HD mice. As we pointed out above, the balance of activation of synaptic and extrasynaptic receptors plays a role in excitability changes in HD. We also previously demonstrated that abnormal sensitivity to NMDA and Mg2+ indicates that NMDAR alterations occur very early in development and suggest the presence of constitutively abnormal NMDARs (Starling and others 2005). These alterations may contribute to an enhancement of NMDA responses at hyperpolarized membrane potentials that may be a key factor in striatal neuronal dysfunction (Raymond and others 2011).
Intrinsic Striatal Connectivity
Besides changes in glutamatergic inputs, intrinsic synaptic activity in striatum is altered in HD. About 95% of striatal neurons are GABAergic MSNs and the remainder are interneurons. Although representing less than 5% of the entire striatal neuron population, interneurons play critical roles in striatal function. The two main types are the large aspiny cholinergic (ACh) interneurons, also known as tonically active neurons (TANs), and a number of diverse types of GABAergic interneurons (Assous and others 2018; Tepper and others 2018). Several types of striatal interneurons are spared from degeneration in HD (Ferrante and others 1987; Harrington and Kowall 1991). More systematic studies, however, found that at least one class of striatal interneuron, the PV-expressing fast-spiking interneuron (FSI), also degenerates in HD (Reiner and others 2013). Why some types of interneurons are spared and others are not remains unknown. What is known, based on recent studies of striatal interneurons in HD models, is that the interneurons are generally dysfunctional and display both morphological and synaptic adaptations.
GABAergic Interneurons in HD
Progressive changes in excitatory inputs are expected to induce compensatory changes in postsynaptic neurons. In agreement, there is evidence that the excitatory-inhibitory balance also is disrupted in the HD striatum (Cepeda and others 2007; Holley and others 2019b). Surprisingly, in parallel to the progressive decrease in excitatory inputs, spontaneous GABA synaptic activity increases (Centonze and others 2005; Cepeda and others 2004; Dvorzhak and others 2013; Fig. 2). In support of upregulated GABA transmission, the expression of α1 and α3 GABAA receptor subunits is increased (Cepeda and others 2004; Du and others 2017). Interestingly, while phasic GABA synaptic activity is increased in HD mice, tonic GABA activity is significantly reduced, particularly in indirect pathway MSNs, suggesting both upregulation and downregulation of GABA function (Cepeda and others 2013; Dvorzhak and others 2013; Wojtowicz and others 2013). A reduction in δ subunit expression could explain the decreases in tonic GABA function (Du and others 2017; Rosas-Arellano and others 2018).
What could be the reason for these neuroadaptations? One possibility is that, as a response to early increases in glutamate release in the corticostriatal pathway (Cepeda and others 2003; Joshi and others 2009), inhibitory synaptic activity compensates and becomes enhanced in order to reduce striatal hyperexcitability by shunting excitatory inputs in MSNs or by acting on presynaptic GABAB receptors on corticostriatal terminals to reduce glutamate release (Logie and others 2013). This does not explain, however, why GABA synaptic activity remains increased in spite of progressive corticostriatal disconnection. GABA itself can be excitatory in normal and disease conditions (Bracci and Panzeri 2006). In HD models, several studies have shown excitatory effects of GABA as a result of reduced expression of the chloride transporter KCC2 (Dargaei and others 2018; Hsu and others 2018).
The most likely potential explanation for the origin of increased GABA synaptic activity is that it involves multiple sources (Cepeda and others 2013). Our recent studies showed that somatostatin (SOM)-expressing interneurons (low-threshold spiking, LTS) are prime candidates for increased GABA synaptic activity as they fire more in HD mice (Cepeda and others 2013; Holley and others 2019b). FSIs displayed a significant loss of dendritic complexity, reduced cell membrane capacitance, increased input resistance, and more depolarized resting membrane potentials, suggesting increased excitability with disease progression (Holley and others 2019a). In agreement, optogenetic stimulation of PV-expressing interneurons in R6/2 mice induced significantly larger amplitude MSN responses in HD compared with WT mice (Cepeda and others 2013).
MSN-MSN Connectivity
Another possible source of increased GABA synaptic activity is the MSNs themselves. MSNs are GABAergic and are interconnected via axon collaterals (Czubayko and Plenz 2002) as well as projecting outside the striatum. Similar to FSIs, they undergo significant morphological changes with disease progression including loss of dendritic spines, reduced somatic area, and decreased dendritic field complexity (Klapstein and others 2001). Also like FSIs, the membrane input resistance of MSNs increases and their resting membrane potential becomes depolarized. This enhanced intrinsic excitability could favor increased GABA synaptic activity (Cepeda and others 2013; Dvorzhak and others 2013). We examined MSN-MSN connectivity in HD mice. Dual patch recordings between MSNs demonstrated reduced connectivity between MSNs. However, while connectivity was strictly unidirectional in controls, in HD mice bidirectional connectivity occurred perhaps as a compensatory response (Cepeda and others 2013).
ACh Interneurons
Reduced ACh levels are found in HD human patients and in animal models (Smith and others 2006). Although the number of ACh interneurons is not reduced in HD mouse models, they do undergo morphological and functional alterations (Deng and Reiner 2016; Holley and others 2015; Tanimura and others 2016). Immunocytochemical analysis demonstrated that somatic areas are significantly smaller in symptomatic R6/2 mice compared to WT littermates and, consistent with reduced somatic size, ACh interneurons display smaller membrane capacitance and higher input resistance (Holley and others 2015). Importantly, rhythmic firing of these interneurons was disrupted, probably as a consequence of increased inhibitory inputs from SOM-expressing interneurons. In the Q175 model, a deficit in thalamostriatal transmission onto ACh interneurons was found (Tanimura and others 2016). In contrast, a facilitation of corticostriatal transmission occurred simultaneously and was due to postsynaptic upregulation of voltage-dependent Na+ channels, without detectable changes in glutamate release probability (Fig. 3).
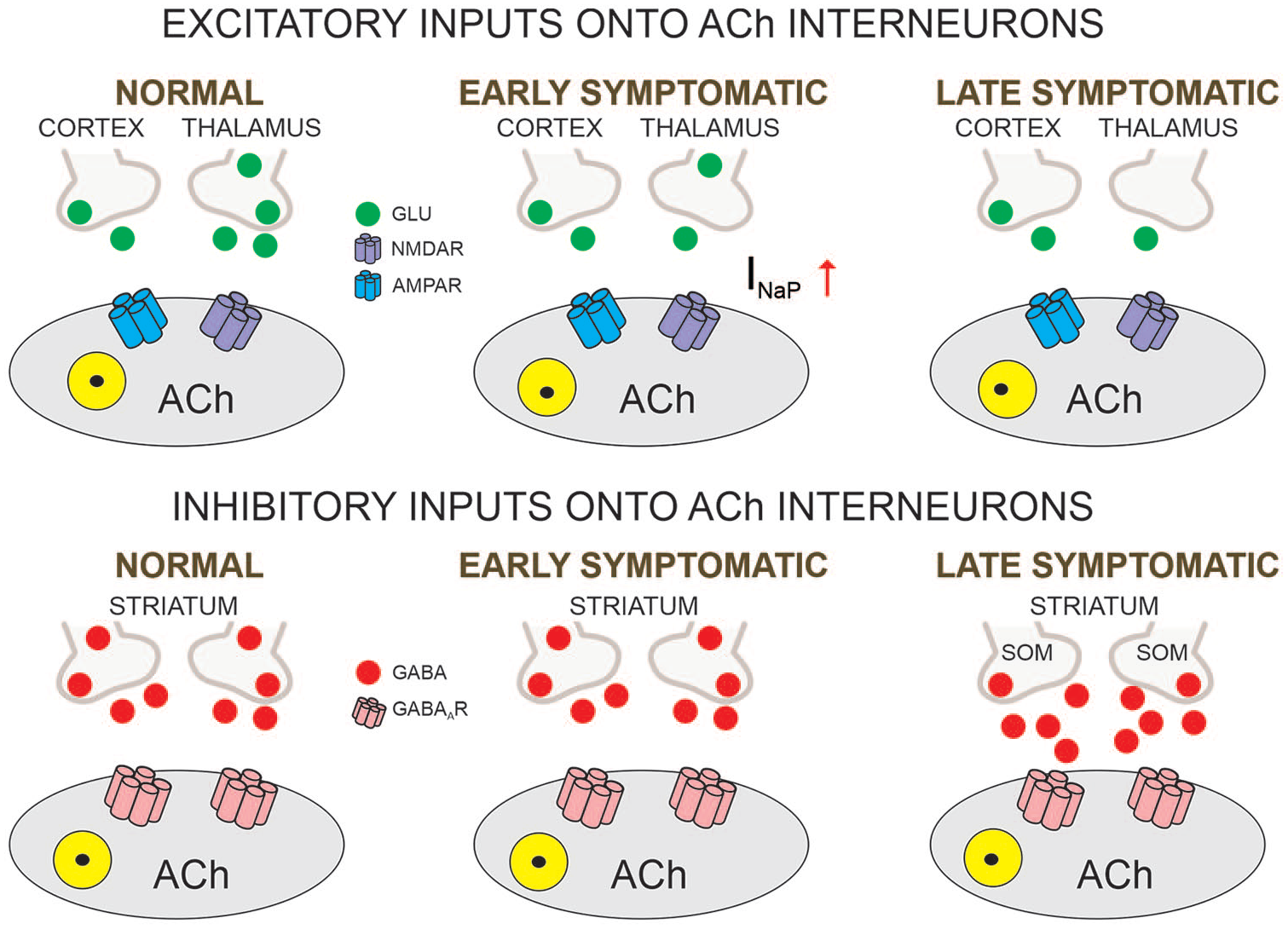
Glutamatergic and GABAergic inputs onto ACh interneurons. The main excitatory input onto cholinergic (ACh) striatal interneurons comes from the thalamus. Reduced inputs trigger compensatory postsynaptic changes in the persistent Na+ current, with consequent upregulation of corticostriatal inputs but no detectable changes in glutamate release probability. In contrast, GABA synaptic activity is increased in the late symptomatic stage, mainly mediated by increased activity of striatal somatostatin (SOM)-expressing interneurons.
Striatal Output Structures
MSNs originate two output pathways, the direct and the indirect pathways. Direct pathway MSNs innervate the SNr and express D1 dopamine (DA) receptors while the indirect pathway MSNs project to the GPe and express D2 DA receptors. The GPe, in turn, innervates the subthalamic nucleus (STN) before reaching the SNr (Plotkin and Goldberg 2019). The two pathways interact to facilitate (direct pathway) or impede (indirect pathway) movement (Box 1). It is believed that in HD indirect pathway MSNs are lost before those of the direct pathway (Deng and others 2004; Reiner and others 1988). Altered membrane and synaptic properties of MSNs in HD predict changes in GPe and SNr. However, it is difficult to predict the overall effect on output regions. On one hand, increased MSN excitability would predict enhanced responses. On the other hand, at least in mouse models, because of increased GABA synaptic activity in the striatum one could expect reduced MSN output.
External Globus Pallidus and Subthalamic Nucleus
In the GPe, two principal types of neurons can be distinguished based on firing properties and the presence or absence of hyperpolarization-activated currents (Ih). The most prevalent, type A (also known as type I or prototypical), is GABAergic, expresses PV, projects to the STN, has a high firing rate, and displays a prominent Ih. Type B (also known as type II or arkypallidal) represents one third of GPe neurons, is also GABAergic, expresses Npas1 and Foxp2, projects to the dorsal striatum, fires at slower rates than type A, and has no Ih (Hegeman and others 2016). In symptomatic R6/2 mice, cell membrane capacitance and input resistance of type A, prototypical neurons were increased compared with controls. In addition, these changes were accompanied by altered firing patterns including increased interspike interval variation, reduced number of spikes evoked by depolarizing current pulses, but increased number of bursts. In addition, blockade of GABAA receptors facilitated bursting activity of these neurons in R6/2 mice, probably caused by a combination of altered intrinsic and synaptic membrane properties (Akopian and others 2016). Recently, electrophysiological/optogenetic studies examined responses evoked in GPe neurons by selective stimulation of indirect pathway MSNs in R6/2, YAC128, and Q175 models (Barry and others 2018; Perez-Rosello and others 2019). In both studies an increase in GABA current charge was observed, albeit in one study this was caused by a longer response decay time without changes in amplitude (Barry and others 2018), whereas in the other it was caused by an increase in amplitude of the evoked response but no changes in decay time (Perez-Rosello and others 2019). Interestingly, this latter study showed that optogenetic stimulation of indirect pathway MSNs inhibited GPe neuron firing for longer periods of time in Q175 mice compared to WTs, suggesting that GABA may linger in the synaptic cleft (Fig. 4). Further studies in our laboratory aimed at understanding potential mechanisms revealed that the expression of GAT-3, an astrocytic GABA transporter predominant in GPe, is significantly reduced, which could explain longer pauses in GPe neuron firing as well as longer response decay times (Barry and others 2020). Alterations in GABA receptor signaling also were found in the GPe of the R6/1 mouse model at presymptomatic and symptomatic stages. Western blot and qPCR analyses showed that the expression of proteins involved in pre- and postsynaptic membranes, including α1, β2, and γ2 subunits of the GABAA receptor, GAD 65 and 67, the vesicular GABA transporter VGAT, and gephyrin, were significantly decreased. Additionally, a reduction in the frequency of spontaneous and miniature IPSCs was observed (Du and others 2016). This probably indicates that increased charge of evoked GABA responses is more likely due to reduced function of astrocytic GABA transporters, as we recently reported (Barry and others 2020) rather than or in addition to a change in proteins involved in synaptic membranes.
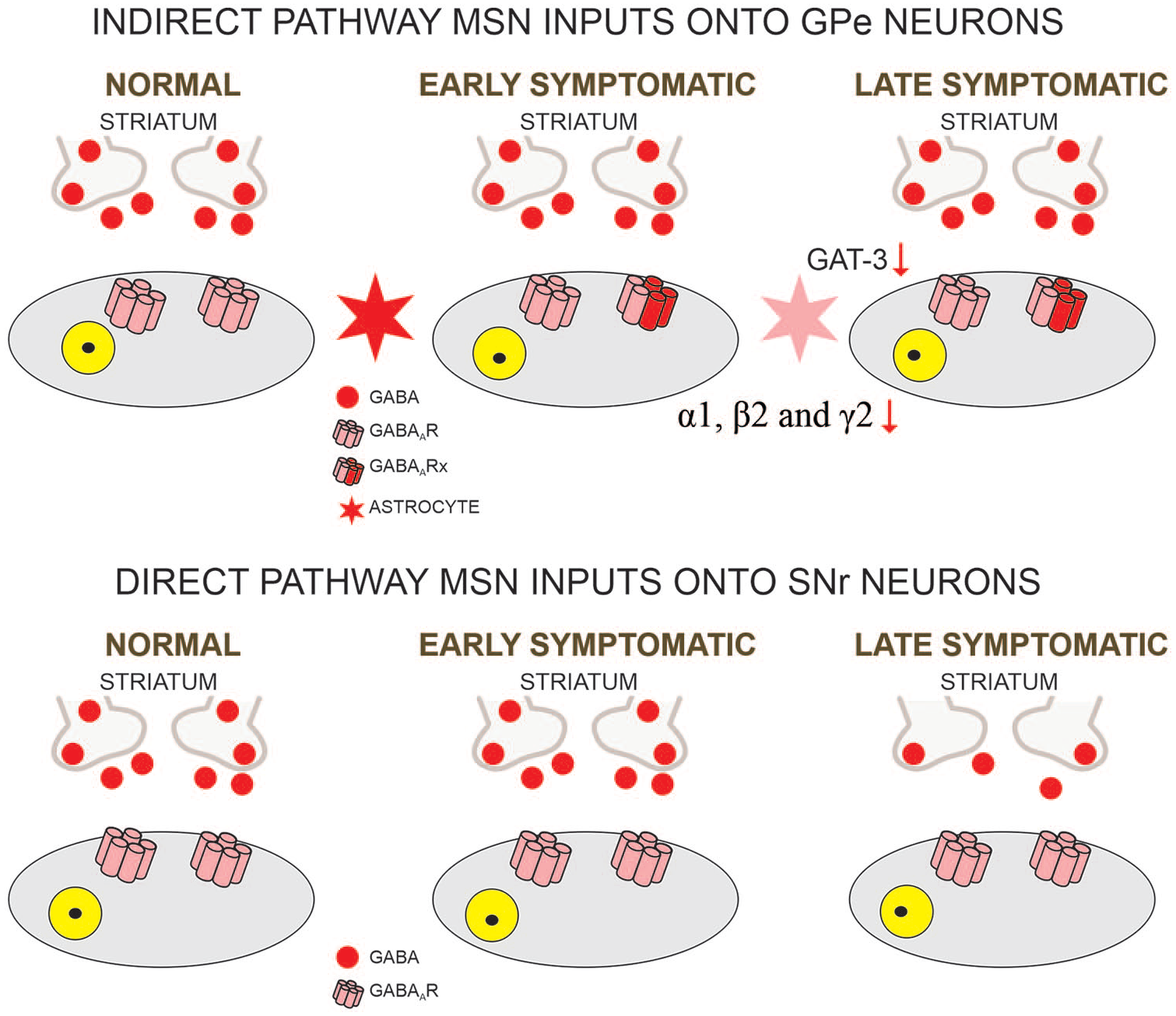
MSN GABAergic inputs onto output regions. Although GABA release from indirect pathway MSNs does not appear affected in HD, changes in GABA receptor subunits can be observed, along with reduced expression of GABA transporter GAT-3. This facilitates postsynaptic responses in GPe neurons. In the late stages of HD progression, the input of SNr/GPi neurons is significantly reduced. MSN = medium-sized spiny neuron; HD = Huntington’s disease.
In the tgHD rat model, reduced firing frequency of GPe neurons occurred, along with increased firing frequency in the STN (Vlamings and others 2012), which would agree with increased inhibition of GPe neurons leading to disinhibition of STN neurons and motor impairment. In YAC128 mice, biphasic changes in excitability and cortical entrainment of STN activity (the cortico-STN hyperdirect pathway) were observed. In the early stages, both the cortex and the STN exhibited neuronal hyperexcitability. In the late stages cortical entrainment of STN activity was disrupted and the spontaneous discharge of STN neurons decreased, suggesting progressive disconnection of the hyperdirect pathway (Callahan and Abercrombie 2015). In BACHD and Q175 mice at 2 and 6 months autonomous STN activity was impaired due to activation of KATP channels. STN neurons also displayed prolonged NMDAR-mediated synaptic currents, caused by a deficit in glutamate uptake and elevated mitochondrial oxidant stress (Atherton and others 2016).
Substantia Nigra Pars Compacta (SNc)
The SN plays an important role in HD pathophysiology. One of the primary causes of chorea is believed to be an excess of DA release onto striatal MSNs (Cepeda and others 2014). In agreement, depletion of brain monoamines with tetrabenazine (TBZ), a vesicular monoamine transporter 2 inhibitor, is commonly used to treat chorea (Huntington Study Group 2006). However, with disease progression DA levels diminish drastically leading to bradykinesia and rigidity. Biphasic changes in DA are reminiscent of biphasic changes in glutamate transmission. The origin of early increases in DA is unknown but initial loss of discrete groups of MSNs in the striatal patch (striosome) compartment projecting to the SNc could contribute to upregulation of DA release. For example, anatomical studies have suggested that early degeneration of striosomal MSNs may produce hyperactivity of the nigrostriatal dopaminergic pathway, causing chorea and other early clinical manifestations of HD (Hedreen and Folstein 1995). It is important to note that increases in DA release in rodent HD models have been observed only rarely, which may provide an explanation why most animal models do not typically show choreiform movements. Nonetheless, in some rodent models upregulation of DA function has been demonstrated. For example, in a rat model of HD (tgHD) that displays hyperkinesia and chorea sensitive to TBZ treatment, there is an increased number of DA cells in the SNc (Jahanshahi and others 2010; Zeef and others 2014). YAC128 mice in the early stages show increased stereotypies that are decreased by TBZ treatment, supporting increased glutamate activity and DA tone in direct pathway neurons (Andre and others 2011). In the R6/2 model, which better replicates the juvenile form of HD characterized by akinesia, rigidity, and epileptic seizures, biphasic changes in DA activity have been more difficult to establish. However, some studies have described hyperactivity in the open field at 3 weeks of age, followed by hypoactivity, starting at 5 weeks (Bolivar and others 2003; Luesse and others 2001), thus replicating human HD progression. We are not aware of DA measurements at 3 weeks of age. However, in presymptomatic R6/2 mice (4 weeks of age) tyrosine hydroxylase (TH, the rate-limiting enzyme for DA biosynthesis) activity was increased while protein levels remained unchanged (Yohrling and others 2003).
Many studies have confirmed progressive loss of DA in the striatum in HD animal models (Johnson and others 2006; Ortiz and others 2011; Ortiz and others 2012). This deficit, along with the progressive corticostriatal disconnection, could explain the hypokinetic phenotype. In the slowly progressing R6/1 model, biphasic changes in DA function are more apparent. Although DA neurons in the SNc did not show changes in basic membrane properties, there was a selective loss of small-conductance Ca2+-activated K+ channels (SK3, responsible for the slow after-hyperpolarization), leading to hyperexcitability and concomitant increases in DA release followed by drastic reductions in DA availability in late stages of the disease (Dallerac and others 2015). Again, this replicated biphasic changes in DA function in HD (Fig. 5). DA deprivation may lead to deleterious consequences similar to those observed in Parkinson’s disease (PD), including bradykinesia. The progressive loss of DA in HD animal models could explain the increased bursting tendency of striatal ACh and SOM interneurons, as well as GPe principal neurons. Electrophysiologically, loss of DA could favor the occurrence of membrane oscillations and bursting activity. For example, in a model of PD produced by DA-depleting lesions, about half of MSNs (both D1 and D2) in slices generated giant spontaneous, depolarizing GABA currents as a result of burst firing from LTS interneurons but not FSI or ACh interneurons (Dehorter and others 2009).
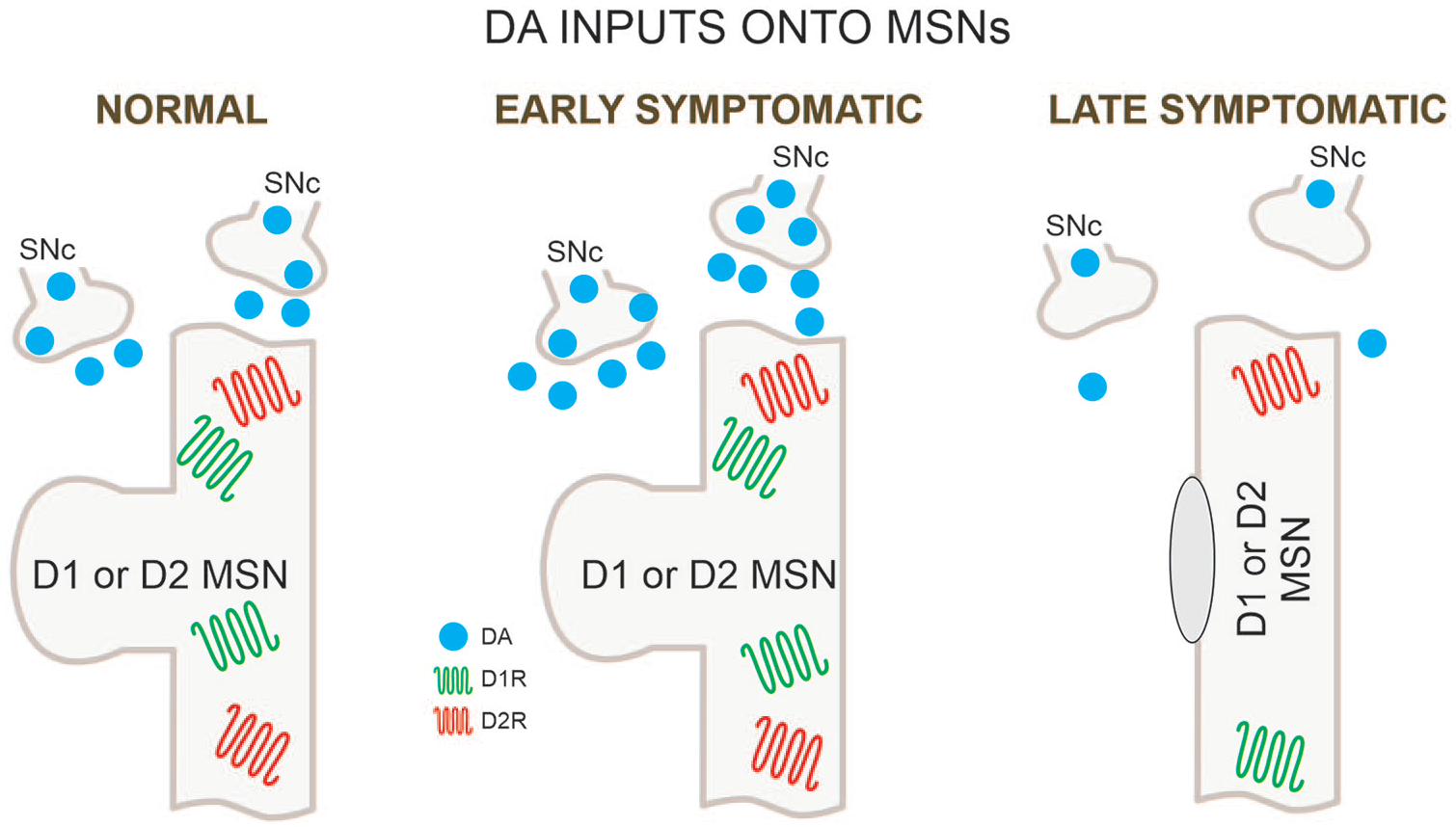
DA inputs onto MSNs. DA inputs from the substantia nigra pars compacta (SNc) undergo biphasic changes with increases followed by decreases. In the late stages DA release is dramatically reduced. MSN = medium-sized spiny neuron; DA = dopamine.
Substantia Nigra Pars Reticulata (SNr)
The SNr is the main output nucleus of the basal ganglia. Most cells are GABAergic and regulate motor activity via an inhibitory projection to thalamic nuclei. SNr cells also project to the superior colliculus where they contribute to the regulation of saccadic eye movements (Hikosaka and Wurtz 1983). When the SNr is activated by excitatory inputs from the STN it prevents movement and when inhibited by striatal MSNs of the direct pathway it promotes movement. We examined intrinsic and synaptic changes in SNr of symptomatic R6/2 and presymptomatic or symptomatic YAC128 mice (Barry and others 2018). Cell membrane capacitance was decreased, whereas input resistance was increased in SNr neurons from R6/2, but not YAC128 mice. Notably, the amplitude of GABA responses evoked by optogenetic stimulation of direct pathway striatal terminals was reduced in SNr neurons of symptomatic mice in both models (Fig. 4). In addition, a decrease in spontaneous GABA synaptic activity, in particular large-amplitude events, in SNr neurons also occurred. We are not aware of other electrophysiological studies examining SNr responses induced by direct pathway stimulation in HD models. However, in freely moving CAG140 mice, an increase in burst rate of SNr neurons was observed (Murphy-Nakhnikian and others 2012). Although the mechanism underlying increased bursting is not known, we speculate that reduced striatal input from direct pathway MSNs plays a role. Finally, while increased bursting of SNr neurons could increase inhibition in the thalamus and promote hypokinesia, the behavioral implications remain speculative (Murphy-Nakhnikian and others 2012).
Rescuing Synaptic Dysfunction
Many approaches have been used to target synaptic abnormalities and rescue dysfunction. We will briefly mention just a few of these interventions concentrating on the corticostriatal pathway in HD animal models. These include enhancing trophic support mechanisms, evaluating the role of glia, examining glutamate transporters, examining the role of cholesterol, and replacing neurons with stem cells.
One of the unintended consequences of the progressive corticostriatal disconnection is the reduction in trophic support and alterations in postsynaptic TrkB receptors (Plotkin and others 2014). The main striatal source of BDNF is from CPNs as well as local astrocytes and oligodendrocytes. Many studies have shown a significant reduction in BDNF production and transport onto the striatum of HD models (Gauthier and others 2004; Zuccato and others 2005). As GABAergic synaptic activity in striatal MSNs is upregulated in HD mice, we tested the effects of bath application of BDNF in slices from symptomatic R6/2 mice. Bath application of BDNF significantly reduced the frequency of spontaneous GABAergic synaptic currents in MSNs from R6/2 but not WT mice (Cepeda and others 2004). We also measured the effects of exercise on corticostriatal synaptic function in R6/2 mice because it increases BDNF (Cepeda and others 2010). We found that exercise prevented the reduction in striatal MSN membrane capacitance, but did not reestablish synaptic communication. Interestingly, a recent study directly addressing the corticostriatal disconnection showed that selective optogenetic stimulation of the supplementary motor cortex (M2) to dorsolateral striatum pathway rescued motor learning and coordination deficits in symptomatic R6/1 mice. Remarkably, these effects were associated with improvements in synaptic plasticity and normalization of spine density in striatal MSNs (Fernandez-Garcia and others 2020). Thus, pharmacological, behavioral, and electrical or optical stimulation treatments aimed at restoring corticostriatal connectivity and function may be promising therapeutic avenues in HD.
There also has been a growing interest in the role of glia, particularly astrocytes, in HD (Gray 2019; Khakh and others 2017). A pioneering study of striatal astrocytes in HD mouse models demonstrated that symptom onset is associated with decreased Kir4.1 channel function, which leads to elevated extracellular K+ and increased MSN excitability (Tong and others 2014). Other studies have shown multiple deficiencies in astrocytic function (Dvorzhak and others 2016; Lee and others 2013b). However, it is currently difficult to determine whether astrocyte abnormalities are secondary to neuronal dysfunction or they are independent; most likely they occur in parallel (Al-Dalahmah and others 2020). Nonetheless, using a genetic approach, it was shown recently that lowering mHTT expression from astrocytes improves motor, psychiatric, neuropathological, and electrophysiological phenotypes of BACHD mice (Wood and others 2019). Similarly, using Zinc Finger Proteins (ZFP) to reduce mHTT from astrocytes, gene expression alterations were reversed in Q175 and R6/2 models suggesting that mHTT lowering strategies for the treatment of HD symptoms should target both neurons and glia (Diaz-Castro and others 2019).
The idea that glutamate transport is altered and potentially downregulated in HD has received strong support. Interestingly, ceftriaxone, an antibiotic that increases the glutamate transporter 1 (GLT-1), ameliorated behavioral symptoms in R6/2 mice during the early symptomatic stage (Miller and others 2008). However, the beneficial effects of ceftriaxone are difficult to interpret due to pleiotropic actions such as attenuation of pro-inflammatory mediators and oxidative stress, as well as upregulation of BDNF (Yimer and others 2019). Diminished function of GLT-1 could be associated with astrocyte alterations as studies have shown in HD models that glutamate uptake critically depends on Kir4.1 (Dvorzhak and others 2016). Moreover, imaging of glutamate signals revealed longer duration decay times suggesting deficient glutamate clearance in R6/2 mice (Jiang and others 2016). This finding was corroborated in Q175 mice (Dvorzhak and others 2019). In contrast, other studies using a similar approach found that glutamate clearance in the HD striatum is normal or even accelerated, particularly in the aggressive R6/2 model (Koch and others 2018; Parsons and others 2016). This unexpected finding suggests that glutamate transporter dysfunction is not a major component of HD pathology and casts doubt on the use of glutamate uptake enhancement as a therapeutic strategy for the treatment of HD (Parsons and others 2016). Clearly, more studies on glutamate transport are warranted. In particular, the role of other transporters, for example, GLAST, need to be examined. In CPNs from R6/2 or WT mice, only concurrent, but not individual, inhibition of GLT-1 or GLAST induced a significant increase in EPSC decay time and this effect was significantly greater in R6/2 CPNs. This indicates that neurons in this area may have homeostatic mechanisms that involve upregulation of either transporter when the other is deficient (Estrada-Sanchez and others 2019).
Cholesterol is made in astrocytes and its biosynthesis is perturbed in HD patients and animal models (Valenza and others 2015). Cholesterol dysfunction can affect synaptic transmission in two ways. First, as the main component of myelin, impaired cholesterol production can lead to demyelination (Rosas and others 2006) and thus to impairment of electrical impulse transmission in HD. Reduced myelin is an early pathological event in HD that precedes behavioral abnormalities and neuron loss. Besides myelin formation, cholesterol plays an important role in synaptic transmission per se. For example, the level of cholesterol in the synapse is critical in limiting spontaneous synaptic vesicle fusion and alterations in synaptic cholesterol metabolism can be a critical determinant of glutamatergic neurotransmission (Wasser and Kavalali 2009). Interestingly, BDNF stimulates de novo synthesis of cholesterol in neurons but not in glial cells. It also promotes the development of neuronal lipid rafts (cholesterol/sphingolipid microdomains involved in protein trafficking) and increases the levels of presynaptic proteins necessary for the readily releasable pool of synaptic vesicles (Suzuki and others 2007). As reduced cholesterol levels lead to instability of surface AMPAR and loss of synapses and spines (Hering and others 2003), stimulation of lipid raft formation by BDNF could improve synaptic transmission.
In rodent models of HD, neuronal loss is only mild or occurs very late in disease progression. Thus, symptoms are caused principally by cell dysfunction, in particular abnormal synaptic communication (Levine and others 2004). In contrast, in human patients clinical symptoms occur as a combination of functional and structural changes. One question is how can we replace the massive loss of neurons in the striatum and other brain regions that occurs in HD? Implantations of neuronal cell lines have now been used to treat HD symptoms in animal models (Holley and others 2018). These include adult multipotent stem cells, pluripotent stem cells and, more recently, embryonically derived neural stem cells (NSCs), which can differentiate into neurons, glia, and oligodendrocytes. We used ESI-017-derived human (h)NSCs for transplantation studies in HD mice and demonstrated that hNSCs survive, make synaptic contacts, and are electrophysiologically active in striatum and cerebral cortex of R6/2 mice. They also partially rescue some MSN membrane properties, attenuate epileptiform activity, and improve some behavioral symptoms (Reidling and others 2018). These differentiated stem cells also increase BDNF and reduce aberrant accumulation of mHTT in the striatum of transplanted animals. Thus, hNSCs have the potential of restoring corticostriatal connectivity. A recent study in a rat model of HD also showed that human striatal progenitors undergo maturation, integrate into host circuits, and extend projections to the appropriate target regions (Besusso and others 2020).
Future Directions and Conclusions
Although animal models of HD have provided a wealth of information regarding synaptic pathology, those models hardly recapitulate the cardinal motor abnormality, chorea. Thus, electrophysiological recordings of the corticostriatal pathway and output structures from models that display chorea, for example, BACHD-ΔN17 mice (Gu and others 2015; however, see Goodliffe and others 2020) or aged tgHD rats, should prove invaluable to understand the mechanisms of this cardinal symptom. In terms of underexplored cell types and circuits in HD models, it will be important to examine the role of additional striatal GABAergic types such as calretinin-, NPY-, and tyrosine hydroxylase (TH)-expressing interneurons. More functional studies of the thalamo-cortical pathway as well as the respective role of the patch and matrix compartments in HD also are warranted (Crittenden and Graybiel 2011). In addition, knowing the temporal sequence of pathologic events in HD is of paramount importance. This implies parallel neurodevelopmental morphological and functional studies in cortical and basal ganglia structures (Cepeda and others 2019). In a sense, neurodegeneration tends to revert the brain to an immature state. For example, it has been shown that mHTT redirects an intracellular store of juvenile NMDARs containing GluN3A subunits to the surface of striatal neurons leading to abnormally enhanced NMDA currents (Marco and others 2013). Genetic deletion of GluN3A prevents synapse degeneration, ameliorates motor and cognitive decline, and reduces neuronal loss in YAC128 mice. Other studies have shown that depolarizing, potentially excitatory actions of GABA, typically seen during early brain development, reoccur in the hippocampus of R6/2 mice and are caused by a decrease in KCC2 expression and an increase in NKCC1 (Dargaei and others 2018). Furthermore, the NKCC1 inhibitor bumetanide abolished the excitatory action of GABA and rescued the performance of R6/2 mice on hippocampal-associated behavioral tests. Considering the critical role of normal HTT in cortical development reviewed earlier, it is relevant to mention a recent study in a mouse model of cortical injury. Tuszynski and colleagues discovered that after injury, the regenerative transcriptome of mature corticospinal tract motor neurons reverts back to an embryonic state. Surprisingly, the authors also found that one of the essential genetic pathways promoting neuronal growth and repair involves the HTT gene (Poplawski and others 2020).
In terms of therapies, strategies for lowering mHTT appear very promising (Li and others 2019; Tabrizi and others 2019; Zeitler and others 2019). However, most therapeutic trials aimed at lowering mHTT are performed when dysfunction is already present. If future studies confirm that HD is not only a neurodegenerative but also a neurodevelopmental disease, only by targeting early brain development can therapies be efficient. For example, using human HD induced pluripotent stem cell cultures it was demonstrated that the presence of mHTT negatively affects striatal and cortical neuronal progenitor specification and commitment. These defects could be rescued by down-regulating mHTT with synthetic ZFPs (Conforti and others 2018). A recent study using a disease-on-a-chip microfluidic platform to examine the corticostriatal network in vitro, provided further evidence that cortical alterations are critical to the progression of the disease and that substitution of HD cortical neurons with WT neurons was sufficient to rescue cellular alterations in mutant striatal neurons (Virlogeux and others 2018).
Finally, the role of microglia in HD pathology deserves more attention. Indeed, microglia may play an intimate role in synaptic alteration and loss during HD pathogenesis (Savage and others 2020). In this study, the maturation of striatal microglia was examined using anti-IBA1 staining in the R6/2 model at 3, 6 and 13 weeks of age. At 3 weeks, microglia from R6/2 mice displayed a more mature morphological phenotype, as demonstrated by increased somatic and arborization areas compared to those from WT mice. In addition, microglial processes from 10-week-old R6/2 mice made fewer contacts with synaptic structures than microglial processes from 3-week-old R6/2 mice and age-matched WT littermates. Across all ages, R6/2 microglia also demonstrated increased phagocytosis. In another study, a subtype of reactive astrocytes present in HD patients, called A1, was induced by activated neuroinflammatory microglia. A1 astrocytes were shown to lose the ability to promote neuronal survival and induced the death of neurons and oligodendrocytes (Liddelow and others 2017). Although a recent study found little evidence for the presence of pathogenic A1 astrocytes in HD (Diaz-Castro and others 2019), depletion of microglia from R6/2 mice with the small-molecule receptor tyrosine kinase inhibitor, pexidartinib (PLX3397) reduced grip strength and object recognition deficits, mHTT accumulation, astrogliosis, and striatal volume loss (Crapser and others 2019).
Almost 15 years ago we provided evidence that in HD there is a progressive disconnection in the corticostriatal pathway, which could lead to abnormal sensitivity of postsynaptic glutamate receptors and eventually cell degeneration (Cepeda and others 2007). Alterations in glutamate release and receptor abnormalities may be enhanced by a progressive loss of synaptic communication leading to activation of extracellular NMDARs and a functional decrease in trophic factors (Gladding and others 2014; Raymond and others 2011). In that sense, HD is not exactly a “synaptopathy” but an “extrasynaptopathy.” As pointed out in the hippocampus, activation of synaptic NMDARs is neuroprotective whereas activation of extrasynaptic NMDARs activates cell death pathways (Hardingham and Bading 2010). The hypothesis of progressive disconnection or miscommunication between cortex and striatum is now widely accepted and, we believe, can be generalized to other pathways and regions throughout the cerebral cortex and basal ganglia in HD. The cause of synaptic disconnection may have its roots in the way mHTT interacts with synaptic proteins as well as with voltage- and ligand-gated channels. An aberrant interaction could lead to gain- and loss-of-function. It is tempting to speculate that biphasic changes in glutamate and DA release reflect initial gain-of-function, which consequently engages compensatory neuroadaptations leading to loss-of-function. This could also explain contrasting symptomatology in early and late stages of the disease. Finally, the contribution of non-neuronal elements to HD pathology should not be neglected and, in fact, the evidence now demonstrates they play an active role in synapse dysfunction and loss. Genetic models of HD have taught us invaluable lessons to understand mechanisms and to design rational therapeutic approaches. In particular, we have come to the realization that correcting synaptic dysfunction well before neuronal degeneration occurs is probably the key for the development of successful therapies in HD.
Footnotes
Acknowledgements
We appreciate fruitful and insightful discussions and contributions from present and past members of our laboratory.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was possible thanks to the support of NIH grants NS41574 (MSL), NS096994 (MSL), U54HD087101 (MSL), NS111316 (CC), CHDI Foundation (Contracts A5666, A8462), the Hereditary Disease Foundation, the California Institute for Regenerative Medicine (CIRM) TR2-01841, and Preclinical Grant PC1-08117.