
Abstract
Alzheimer’s disease (AD) is a debilitating age-related neurodegenerative condition. Unbiased genetic studies have implicated a central role for microglia, the resident innate immune cells of the central nervous system, in AD pathogenesis. On-going efforts are clarifying the biology underlying these associations and the microglial pathways that are dysfunctional in AD. Several genetic risk factors converge to decrease the function of activating microglial receptors and increase the function of inhibitory receptors, resulting in a seemingly dampened microglial phenotype in AD. Moreover, many of these microglial proteins that are genetically associated with AD appear to interact and share pathways or regulatory mechanisms, presenting several points of convergence that may be strategic targets for therapeutic intervention. Here, we review some of these studies and their implications for microglial participation in AD pathogenesis.
Alzheimer’s Disease
Alzheimer’s disease (AD) is a debilitating age-related neurodegenerative condition. It is a progressive disorder that deteriorates memory and cognition and is ultimately fatal, with an immense personal and societal burden. Five million individuals in the United States have AD, and this number is projected to increase to about 13 million by 2050 (Brookmeyer and others 2011). Currently, there is no disease-modifying therapy for AD. Over the past few decades, the aging and dementia research community has made progress in identifying risk factors, particularly genetic loci, and defining the clinical and pathologic features of the disease (Morris and others 1993). These discoveries have led to the understanding that AD is a progressive disease with dementia only resulting in its terminal stage. Initially, AD has a long subclinical phase in which molecular changes occur, including the gradual accumulation of characteristic amyloid and tau pathologies, in the absence of overt cognitive impairment. This is followed by a phase of mild cognitive impairment and finally by AD dementia (Sperling and others 2011).
Late-onset Alzheimer’s disease (LOAD), in which disease presents after age 65, accounts for the majority of AD cases. Early-onset Alzheimer’s disease (EOAD) occurs in 5% of cases, in which disease presents before age 65 (Alzheimer’s Association 2015). These early presentations include autosomal dominant AD (ADAD) cases, a rare form of AD in which the disease is inherited in an autosomal dominant fashion at an early age (Bateman and others 2012). ADAD progresses rapidly and is usually associated with mutations in the APP or PSEN gene (Bateman and others 2012). Conversely, LOAD appears to be a sporadic disease, and its etiology and pathogenesis are poorly understood. While clinical presentations suggestive of Alzheimer’s dementia are classified by assessments of cognitive status, AD continues to be biologically defined by neuropathological hallmarks (Jack and others 2018). The known pathology of AD includes aberrant extracellular amyloid-beta (Aβ) aggregates which form diffuse and neuritic plaques and hyperphosphorylated tau aggregates which form intraneuronal neurofibrillary tangles (Duyckaerts and others 2009). These pathological hallmarks are accompanied by loss of synapses, neuronal death, and gross brain atrophy (Duyckaerts and others 2009). Interestingly, initial AD brains characterized by Alois Alzheimer revealed the presence of glial cells with abnormal morphology (Alzheimer 1907; Alzheimer and others 1995; Graeber and others 1997), which were later shown to be astrocytes and microglia (Maragakis and Rothstein 2006; Mosher and Wyss-Coray 2014). While chronic shifts in inflammation and immune signaling in AD were long considered to be a response to pathology, microglia are now regarded as playing key roles in both disease initiation and progression (Box 1). Here, we will review the current literature studying the impact of LOAD genetic risk-variants on the functional responses of microglia, and discuss the implications of these findings.
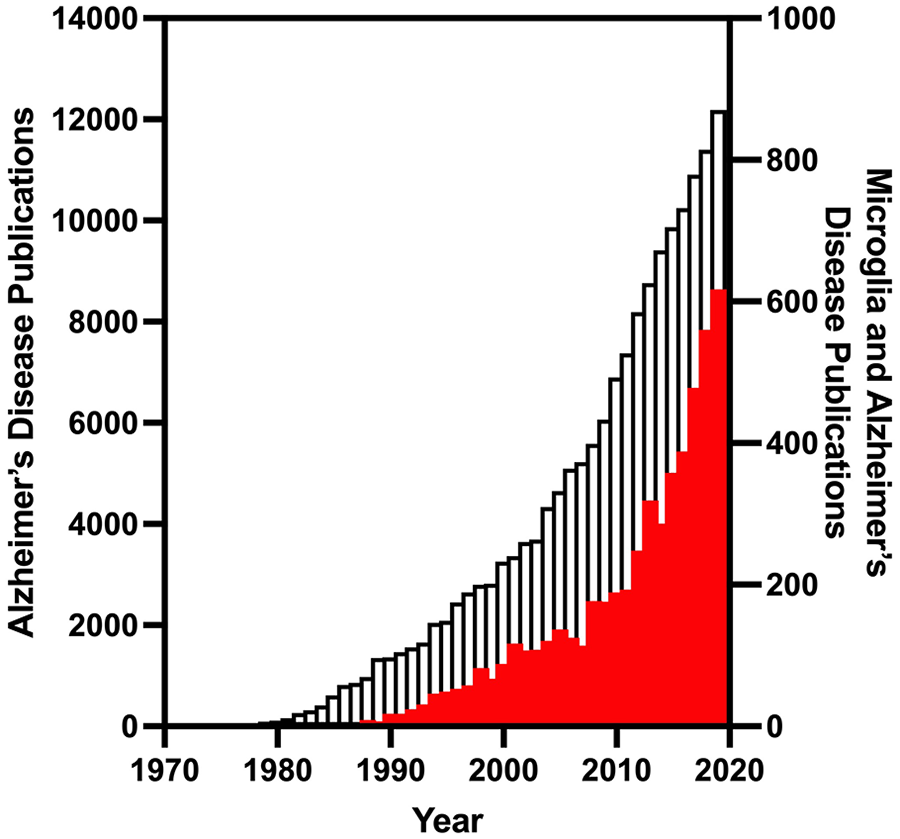
The percentage of Alzheimer’s disease publications that investigate microglia have been increasing over time. Publications listed in PubMed that respond to the search terms “Alzheimer’s disease” or “Alzheimer’s disease AND microglia” between the years of 1970 and 2019 are plotted above. While both search terms have increasing publications over time, the percentage of publications investigating microglia have especially increased in recent years, with a high of 5% in 2019. Open bars correspond to “Alzheimer’s disease” on the left y-axis and red bars indicate “Alzheimer’s disease AND microglia” on the right y-axis.
Microglia
Microglia are the resident innate immune cells of the central nervous system (CNS), where they mediate the local immune response. Like other tissue-resident macrophages, microglia originate from hematopoietic progenitors in the yolk sac, which emigrate to the CNS well before the development of the brain (embryonic days 8.5–10.5 in mice; Alliot and others 1999; Askew and Gomez-Nicola 2018; Ginhoux and others 2010; Ginhoux and others 2013; Hoeffel and others 2012; Schulz and others 2012). Microglia participate in a wide range of homeostatic processes including neurogenesis and synaptic pruning (Hammond and others 2018; Nimmerjahn and others 2005; Schafer and others 2012; Squarzoni and others 2014). They further contribute to maintaining brain homeostasis through constantly surveying the environment and performing innate immune functions such as responding to injury, tissue repair, and removing unwanted molecules or pathogens (Hammond and others 2018; Hanisch and Kettenmann 2007; Nimmerjahn and others 2005).
It has long been accepted and more recently shown that microglia are highly plastic, long-lived immune cells that respond to their environment and modulate their gene expression, phenotype, and function in certain CNS regions or disease states (Mastroeni and others 2017; McCarthy 2017; Olah and others 2018). They are thought to be a heterogeneous population with distinct subsets ranging in function. Indeed, specific microglial subpopulations have been observed in mouse models of Alzheimer’s disease, where microglia were profiled over time at the single-cell level (Keren-Shaul and others 2017; Mathys and others 2017). Microglial heterogeneity is now being explored in the human system by examining single-immune cell transcriptomics from autopsy tissue and biopsy material (Olah and others 2020; Sankowski and others 2019). In one study, the analysis of 21 samples identified nine microglial subsets with varying expressions of activation markers and immune response genes (Olah and others 2020). While the overwhelming majority of microglia belong to clusters that appear to represent homeostatic microglial populations, one subset enriched with markers of microglial activation and antigen presentation was negatively correlated with a clinical diagnosis of AD, AD pathology, and tau tangles. Conversely, a subset featuring genes involved in anti-inflammatory responses (interleukin [IL]-10, IL-4, IL-13) was found to be positively associated with amyloid pathology and clinical AD (Olah and others 2020).
Pre-GWAS Perspective
Prior to the age of transcriptomics and genome-wide association studies (GWAS), microglial profiling in AD was characterized by morphology, targeted phenotyping, and functional outcomes, with an emphasis on inflammatory cytokine production, phagocytosis, and Aβ clearance. Inflammation had long been thought to be a driving force in AD. Several studies including those performed in AD mouse models led to the conclusion that activated microglia contribute to neuronal death in AD, and that they need to be suppressed with anti-inflammatory drugs (Akiyama and others 2000). Aligning with this inflammation theory, epidemiological observations suggested the use of nonsteroidal anti-inflammatory drugs (NSAIDs) to be protective against AD, although a meta-analysis of these findings suggests that the reported benefits of NSAIDs may largely be due to bias in analysis and reporting (de Craen and others 2005; in t’ Veld and others 2001). When tested for preventative effects against AD in a recent clinical trial no evidence was observed for improved outcomes with NSAID treatment in presymptomatic individuals including cognition, and the study raised safety concerns of significant cardiovascular and gastrotintestinal side effects (Meyer and others 2019). The failure of a beneficial effect in clinical trials has contributed to the inflammation dogma being challenged, and it is now suggested that microglial activation may play a beneficial role in AD, as discussed throughout this review. Another clinical trial with ibuprofen and cromolyn has recently been completed, the results of which once published may help clarify whether NSAIDs provide a preventative or therapeutic benefit against AD (Elmaleh 2020).
While innate immune cells have many roles in the CNS, impaired phagocytosis resulting in an inability to clear Aβ has long been thought to be critical to LOAD progression (Fiala and others 2005; Fiala and others 2007; Hickman and others 2008; Wildsmith and others 2013; Yoon and Jo 2012). Aβ clearance is impaired in the AD brain, suggesting an imbalance between rates of Aβ production and clearance (Mawuenyega and others 2010; Wildsmith and others 2013). Interestingly, monocytes and monocyte-derived macrophages from AD subjects are ineffective at phagocytosis of Aβ compared to those from age-matched controls, independent of genetic background (Avagyan and others 2009; Fiala and others 2005; Zaghi and others 2009). As these cells are derived from the circulation, it suggests that there is an underlying defect in the cells themselves, rather than a response to the local AD brain environment. In addition, macrophages from AD subjects appear to undergo apoptosis following exposure to Aβ (Avagyan and others 2009; Fiala and others 2005). These findings suggest that altered phagocytic function of macrophages and microglia contribute to AD pathogenesis. However, the molecular mechanisms underlying impaired phagocytosis and the implications of this altered phenotype for microglial participation in disease is unclear. Genetic studies have begun to clarify risk factors and molecular pathways that may contribute to the mechanisms involved in altered microglial functioning in AD.
GWAS and Sequencing Identification of LOAD-Associated Genes
With the ongoing discovery and validation of LOAD susceptibility loci, genetic risk factors have been identified that give insights into the earliest phases of the pathophysiological processes leading to LOAD (Hollingworth and others 2011a). It is thought that genetics contribute 53% of the phenotypic variance observed in AD (Ridge and others 2016). In particular, unbiased genetic studies have highlighted the role of the innate immune system in LOAD susceptibility, implicating a central role for microglia in disease. Initially, large consortiums such as the International Genomics of Alzheimer’s Project (IGAP) identified several AD-associated genetic loci which implicate genes that are preferentially or exclusively expressed in microglia in the CNS, such as ABCA7, BIN1, CASS4, CD33, CLEF1, HLA, INPP5D, MEF2C, MS4A region, NME8, PICALM, PTK2B, RIN3, SORL1, and ZCWPW1 (Harold and others 2009; Hollingworth and others 2011a; Hollingworth and others 2011b; Lambert and others 2009; Naj and others 2011; Seshadri and others 2010). In the last 7 years, additional GWAS, GWAS by proxy, and sequencing studies have only enhanced the suggestion that genetically susceptible microglia play a central role in LOAD pathogenesis. These genetic findings, in combination with transcriptomic data, have allowed us to hypothesize which microglial functional programs may be impacted in AD. For example, the combined genetic association of CD33, TREM2, TYROBP, and CR1 with AD suggests that phagocytosis may be altered in microglia as a pathogenic mechanism of disease (Bradshaw and others 2013; Griciuc and others 2013; Konishi and Kiyama 2018; Zhang and others 2013).
It is very clear from human genetics and transcriptomics that a microglial-specific intervention could be a strategic therapeutic approach for LOAD (Box 2). However, it may be a major challenge to identify ways in which to positively modulate microglial phagocytic activities while dampening any detrimental properties in order to preserve neuronal function and plasticity. While findings strongly support a causal involvement of microglial cells in AD pathogenesis, the underlying mechanisms remain unclear. GWAS indicate the genetic loci that are associated with disease; they do not definitively identify the associated gene(s). Tools such as fine mapping, eQTL analysis, and other functional genomic studies are needed to elucidate the biology underlying these associations. Such efforts are ongoing and will continue to clarify pathways that are dysfunctional in LOAD (Andrews and others 2020).
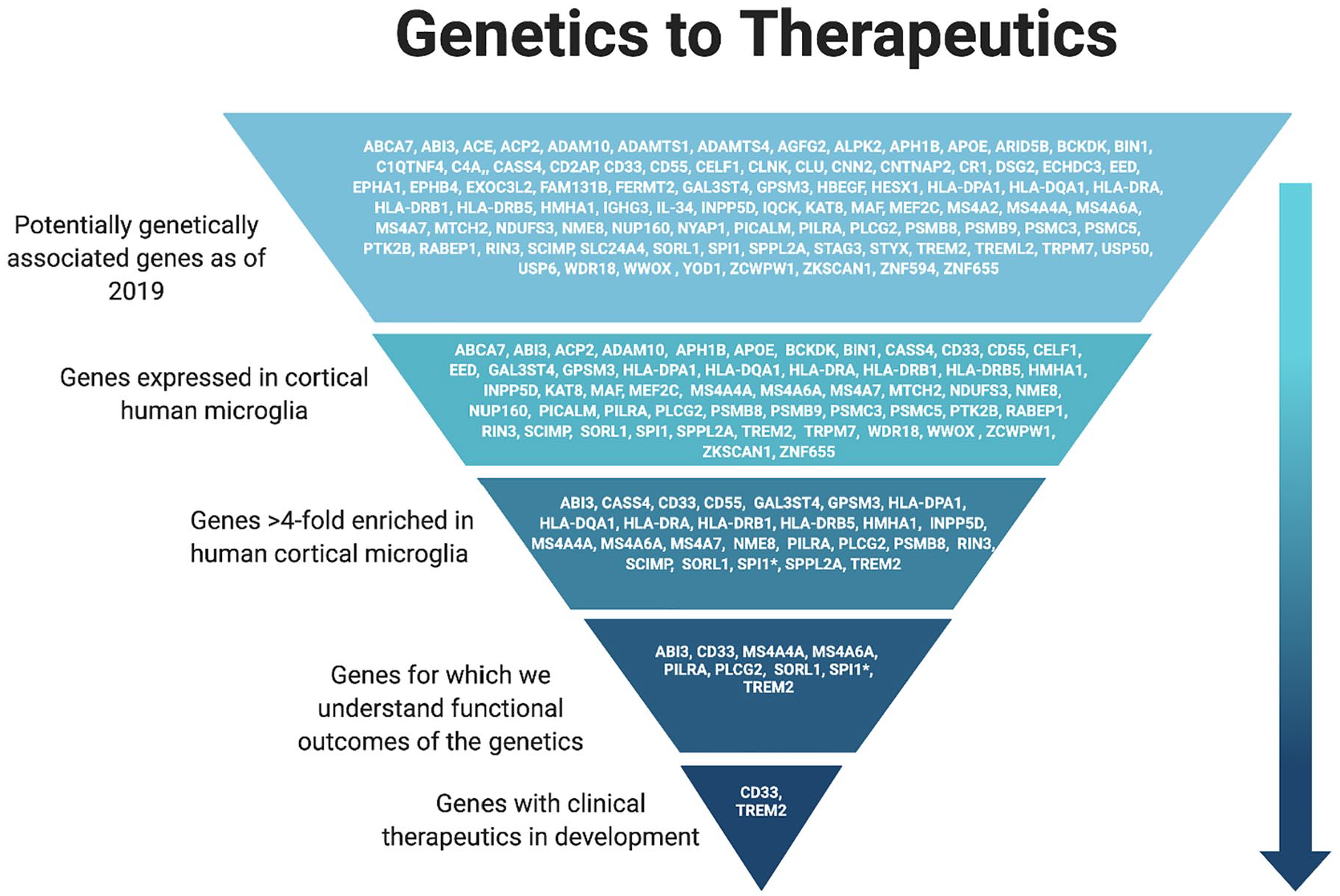
Over the past decade, many genes have been identified as genetically associated with LOAD. Many efforts are underway to identify the cell type(s) affected and the functional outcomes of these associations with the goal of targeting them for treatment of LOAD.
Microglial Suppression in LOAD
The dogma that arose in the pre-GWAS era concluded that the immune system needs to be therapeutically suppressed in AD, and has been supported by studies in AD mouse models (Akiyama and others 2000). However, as reviewed below, the recent human genetic findings are in stark contrast to this hypothesis and conversely suggest that a less responsive microglial phenotype is associated with AD risk. Indeed, AD risk variants appear to converge to suppress immune signaling, while protective alleles enhance the immune response, resulting in a shared inhibitory microglial functional outcome that may contribute to AD pathogenesis (Bradshaw and others 2013; de Rojas and others 2020; Huang and others 2017; Kleinberger and others 2014; Magno and others 2019; Rathore and others 2018; Replogle and others 2015). Microglial suppression resulting in the inability to resolve inflammatory processes, mitigate pathology, and maintain brain homeostasis may heighten susceptibility to neuronal injury and contribute to AD progression. Importantly, this hypofunctional microglial phenotype may extend past genetic risk variants and functionally converge with age-associated senescence, and other lifestyle and environmental factors associated with AD. Below, we review some of these genetic findings and their implications for microglial participation in LOAD pathogenesis. As many of the innate immune proteins genetically associated with LOAD share pathways or regulatory mechanisms, here we additionally outline several points of convergence that may serve as advantageous microglial targets for therapeutic intervention.
CD33
GWAS established the CD33 locus as a genetic risk factor for AD in 2011 (Hollingworth and others 2011b; Naj and others 2011). The locus had been previously identified to be associated with AD in 2008 in a large familial study (Bertram and others 2008). CD33, also known as Siglec-3, is a 67-kDa transmembrane glycoprotein expressed on the surface of myeloid progenitor cells, including monocytes, macrophages, and microglia (Bradshaw and others 2013; Griciuc and others 2013; Malik and others 2013; Walker and others 2015). It functions as a lectin, a carbohydrate-binding protein, and specifically binds sialic acid. CD33 is an inhibitory molecule, limiting endocytosis and the activation of immune signaling pathways, and inducing apoptosis (Crocker and Redelinghuys 2008). Its intracellular component contains immunoreceptor tyrosine-based inhibitory motifs (ITIMs) that are implicated in the inhibition of cellular activity (Crocker and Redelinghuys 2008; Taylor and others 1999). In support of its putative inhibitory function, CD33 has been shown to constitutively repress monocyte-derived pro-inflammatory cytokines (Lajaunias and others 2005). Furthermore, reducing the surface expression of CD33 using siRNA or an anti-CD33 antibody leads to an increase in spontaneous pro-inflammatory cytokine production by monocytes (Lajaunias and others 2005).
There are two main protein isoforms of CD33, termed CD33M and CD33m (Pérez-Oliva and others 2011). The smaller isoform, CD33m, is generated through alternative splicing and lacks the Ig V-set domain which contains the sialic acid-binding motif (Malik and others 2013; Pérez-Oliva and others 2011). This domain is encoded in exon 2. The AD-associated risk allele rs3865444C is strongly associated with increased expression of CD33 exon 2. This can be observed at the protein level with dramatically increased expression of CD33M (the exon 2-containing isoform) with the risk allele (Malik and others 2013; Raj and others 2014). While the function of the small CD33m isoform remains unclear, it has been suggested to be a protective isoform (Estus and others 2019). The causative AD-associated single nucleotide polymorphism (SNP) is most likely rs12459419, which is located in a putative SRSF2 splice site of exon 2 and is in high linkage disequilibrium with the AD-risk GWAS SNP, rs3865444 (Malik and others 2013; Raj and others 2014). CD33 rs3865444C has also been associated with reduced uptake in monocytes (Bradshaw and others 2013). This finding is supported by experiments in which CD33 was expressed in an immortalized microglial cell line and phagocytosis was reduced (Griciuc and others 2013).
The ligands for CD33 include the sialic acid moiety on glycoproteins (Crocker and Redelinghuys 2008). However, few CD33 protein binding partners have been identified, with only CD14 being suggested as a binding partner in monocyte-derived dendritic cells and S100A9 in myeloid-derived suppressor cells (Chen and others 2013; Ishida and others 2014). The binding of CD14 by CD33 is suggested to lead to downregulation of Toll-Like Receptor 4 (TLR4)-mediated signaling (Ishida and others 2014). Upon CD33 signaling, its intracellular ITIM motifs become phosphorylated, which then attract SH2-domain-containing proteins. In the case of CD33, these are primarily thought to be the phosphatases SHP-1 and SHP-2 (Taylor and others 1999). Phosphorylated CD33 has also been reported to recruit the SH2-domain-containing proteins SOCS3 and Cbl (Orr and others 2007; Walter and others 2008). However, it remains unclear which recruited proteins and sialic acid-dependent binding partners are the most relevant in microglia and in the context of AD-associated stimuli such as amyloid pathology.
Of note, the murine ortholog of CD33 lacks the ITIM motif and is expressed only at low levels on microglia, presenting additional challenges in studying the role of CD33 in mouse models of AD (Bhattacherjee and others 2019; Crocker and others 2007). The murine ortholog additionally includes a unique transmembrane lysine residue, whereas the human CD33M contains a unique cytoplasmic tyrosine (Cao and Crocker 2011; Crocker and others 2007). These differences may functionally result in divergent roles for CD33 in regulating phagocytosis in humans compared to mice. While human CD33M impairs phagocytosis in monocytes and primary microglia, murine CD33 has conflicting reports. In one study, primary microglial cultures derived from CD33−/− mice demonstrated increased uptake in vitro (Griciuc and others 2013). However, in a more recent study, no effect of CD33 genetic deletion was found on uptake in the BV-2 mouse microglial cell line, and only deletion of human CD33M, but not murine CD33, resulted in increased uptake in a monocytic cell line (Bhattacherjee and others 2019). Moreover, the protective CD33 rs3865444A variant associated with higher expression of the small CD33m isoform seems to be unique to humans (Schwarz and others 2016). These findings highlight the importance of specifically studying human CD33 in AD pathogenesis. However, in studies in 5xFAD mice, knockout of murine CD33 attenuated amyloid-beta pathology and improved cognition, suggesting the existence of additional mechanisms independent of its ITIM motif or repressed phagocytosis through which CD33 may contribute to AD pathogenesis (Griciuc and others 2019).
As our understanding of CD33 biology and its contribution to AD risk has grown, downregulating its expression or inhibiting its function has become a clear potential therapeutic approach. In fact, clinical trials are currently examining the safety and efficacy of anti-CD33 antibodies as a therapy for AD (Paul 2019).
PILRA
Like CD33, PILRA is a sialic acid-binding and ITIM-containing inhibitory receptor that is specifically expressed in innate immune cells. In the cortex, its expression is enriched 15-fold in microglia compared to bulk tissue (Olah and others 2018). Similar to CD33, its expression was also found to be increased in peripheral leukocytes of AD patients (Park and others 2020). PILRA was determined to be the most likely causal gene in the AD-associated locus originally described as the ZCWPW1 locus (Rathore and others 2018). The PILRA genetic variation leads to a missense amino acid change in the ligand-binding domain, in which the AD-associated risk variant glycine 78 is replaced by an arginine (G78R) in the protective variant. This protective amino acid change from glycine to arginine reduces ligand binding (Rathore and others 2018). As such, like CD33, PILRA is another example of an AD-genetic risk factor that results in the increased function of an inhibitory microglial receptor.
TREM2
Among all the identified AD-associated genetic risk factors involved in innate immune function, none have generated as much translational investigation as the mutations within the TREM2 gene (Guerreiro and others 2013; Jonsson and others 2013). As TREM2 is exclusively expressed in cells of the myeloid lineage, microglia remain a prime focus for TREM2-related AD research (Bouchon and others 2001; Colonna 2003). The identified TREM2 mutations associated with LOAD are primarily within exons and include H15Y, R62H, and R47H. Of these variants, R47H has been the most heavily studied and has been demonstrated to result in reduced ligand-binding affinity and phagocytosis (Kleinberger and others 2014; Yao and others 2019). Similarly, in vitro studies have demonstrated that these mutations reduce lipid–ligand affinity and subsequent signaling, indicating a loss of TREM2 function (Kober and others 2016; Sudom and others 2018; Wang and others 2015). Thus, it is suggested that impaired signaling due to a partial loss of TREM2 function may be a major contributor to AD pathogenesis in individuals with these mutations. Additionally, soluble TREM2 in the CSF of AD patients positively correlates with both total tau and phosphorylated tau, but not with Aβ levels (Piccio and others 2016). Individuals carrying the R47H TREM2 variant have been shown to have increased levels of total and phosphorylated tau, with no effect on amyloid (Lill and others 2015). These findings suggest that the participation of TREM2 in AD may involve tau dysfunction.
The TREM2 signaling partner, TYROBP (also known as DAP12), contains an immunoreceptor tyrosine-based activation motif (ITAM) and is an adaptor protein known to associate with many cell surface molecules (Lanier 2009). TYROBP was identified via an integrated systems approach as a key driver for developing LOAD (Zhang and others 2013). Upon ligand binding of TREM2, TYROBP becomes phosphorylated at its ITAM motif. Syk kinase is then recruited to further initiate downstream signaling, promoting cell survival, proliferation, inflammation, and phagocytosis (Konishi and Kiyama 2018). SHIP1, another AD genetically associated protein (gene name INPP5D), has been shown to inhibit this signaling pathway (Peng and others 2010). INPP5D expression was found to be increased in the brains of LOAD patients and to correlate with amyloid plaque density (Tsai and others 2021). However, further research must be done to uncover the functional outcome of the INPP5D AD-associated variant in microglia, and how its inhibitory effects on the TREM2 signaling pathway impact AD risk.
While TREM2 signaling results in cellular activation, TREM2 is thought to be a negative regulator of the inflammatory response. Unlike other TYROBP-associated activating receptors such as TREM1, antibody crosslinking-induced TREM2 signaling does not result in degradation of IkB-α and nuclear translocation of NF-κB (Bouchon and others 2001). Silencing TREM2 expression with shRNA increased TNF (tumor necrosis factor) production in response to diverse TLR ligands including zymosan and CpG in bone marrow- derived macrophages (BMDM; Hamerman and others 2006). Similar findings have also been demonstrated in BMDM from TREM2 deficient mice, in which increased IL-6 and TNF were observed in response to an LPS challenge compared to wild-type controls (Turnbull and others 2006). Conversely, TREM2 overexpression in microglia has been shown to decrease TNF transcription following exposure to apoptotic neurons (Takahashi and others 2005). These findings suggest a role for TREM2 as a regulator of cytokine production and inflammatory responses in a context-specific manner.
Many functional consequences of altered TREM2 expression have been reported, including impaired phagocytosis. Microglia in TREM2-deficient mice were shown to have reduced expression of CD68, a phagocytic marker, as well as to associate less with dying cells in a mouse model of demyelination (Kawabori and others 2015; Sieber and others 2013). Similarly, TREM2 deficiency in an AD mouse model was shown to reduce internalization of Aβ throughout disease (Jay and others 2017). Expression of a LOAD variant of TREM2 in the BV2 mouse microglial cell line resulted in impaired phagocytosis (Kleinberger and others 2014). A more recent study based on stem cell derived microglia-like cells also found TREM2-deficient microglia to be less effective in Aβ clearance (Claes and others 2019). In this regard, the TREM2/TYROBP signaling pathway presents a potential axis to modulate microglial protective functions, including Aβ clearance (Schlepckow and others 2020).
Mouse models of amyloidosis suggest a protective function for TREM2 in disease pathogenesis. In mouse models of AD crossed with TREM2 or TYROBP knockout mice, microglial activation and Aβ clearance are impaired (Wang and others 2015; Wang and others 2016; Yuan and others 2016). Additionally, recent studies in mouse models of AD demonstrate that overexpression of human TREM2 modulates microglial function and morphology, and improves memory and AD pathology (Lee and others 2018). In mice, Trem2-null microglia also fail to acquire an activated transcriptional signature upon various neuropathological insults (Keren-Shaul and others 2017). However, while loss of TREM2 expression results in a detrimental phenotype in mouse models of amyloidosis, in tau models TREM2 deletion as well as mutant TREM2 appears to be beneficial and protects against neurodegeneration at advanced stages of the disease (Gratuze and others 2020; Leyns and others 2017; Sayed and others 2018). This pathology-specific discrepancy highlights two shortcomings in the field: (1) that we are lacking a robust mouse model of LOAD that encapsulates both amyloid and tau pathologies and (2) that we must comprehensively phenotype individuals selected for clinical trials and treatment to better understand AD stage or pathology-specific effects of therapeutic manipulation.
In fact, clinical trials are currently examining the efficacy of a TREM2 agonistic antibody as a therapeutic approach for AD (Alector-Inc. 2020). This antibody binds TREM2 and activates its signaling, resulting in increased phosphorylation of the downstream effector Syk. In early clinical trial findings, soluble TREM2 was decreased in the cerebrospinal fluid (CSF) following treatment, while a colony-stimulating factor 1 receptor (CSF1R) fragment was increased in the CSF (Wang and others 2020). This therapy aims to increase microglial activation via TREM2 signaling, and may compensate for the reduced microglial responsiveness associated with the TREM2 risk variant, and perhaps the other AD variants discussed in this review. However, close examination for phase and pathology specific effects will be necessary to determine whether activating TREM2 may be beneficial at different stages of the disease.
Interestingly, the CD33 and TREM2 genetic associations converge such that CD33 modulates TREM2 expression, and TREM2 functions downstream of CD33. In a targeted study, the CD33 LOAD risk variant was found to be associated with increased TREM2 surface expression. This association was greatly reduced after adjusting for membrane-bound CD33 expression (Chan and others 2015). Importantly, the CD33 risk variant is associated with seven-fold increased CD33 surface expression (Bradshaw and others 2013). These findings suggest that the degree of membrane-bound CD33 expression mediates the interaction between the CD33 risk variant and TREM2 expression. Additionally, TREM2 has been demonstrated to function downstream of CD33. While knockout of CD33 in a mouse model of AD was found to mitigate pathology, this effect was abrogated upon subsequent knockout of TREM2 (Griciuc and others 2019). Similarly, changes in gene expression observed in CD33 knockout AD mice were found to be dependent on TREM2. The convergence of TREM2 and CD33 highlights the relevance of these two signaling pathways in facilitating LOAD risk, and offers a potentially advantageous point for therapeutic modulation.
TREM1
While not a GWAS association, an intronic variant of TREM1 (rs6910730) that confers AD risk was identified in a targeted analysis of the TREM locus in relation to features of AD (Replogle and others 2015). This variant has been found to decrease TREM1 surface expression in myeloid cells, and is associated with increased neuritic and diffuse plaques, Aβ density, and cognitive decline. TREM1 has been shown to facilitate Aβ phagocytosis (Jiang and others 2016). As this intronic variant is associated with amyloid pathology but not tau pathology, TREM1 may be important in the interaction between Aβ and the innate immune system.
In addition to decreasing TREM1 surface expression, the AD-associated TREM1 intronic variant also decreases the ratio of TREM1:TREM2 expression in myeloid cells (Chan and others 2015). In contrast, another TREM1 variant (rs2627567) that decreases TREM1 expression but does not alter the TREM1:TREM2 ratio was not associated with AD. This finding suggests that the TREM1 association with AD may be mediated not by a simple reduction in TREM1 expression but by influencing the balance of signaling molecules that affect myeloid activation (Chan and others 2015). This concept aligns with the recurring theme of an altered balance of activating and inhibitory signaling molecules in the AD-associated risk variants discussed thus far. Importantly, TREM1 and TREM2 both signal through the adaptor protein TYROBP (Arts and others 2012; Bouchon and others 2001). While crosstalk between TREM1 and TREM2 has not been thoroughly explored, TREM2 appears to negatively regulate the TREM1 proinflammatory response, such that TREM2 signaling reduces TREM1-induced TNFα production (Takahashi and others 2005). This finding highlights the importance of a balanced TREM1:TREM2 ratio for myeloid cell signaling.
The myeloid literature is clear in reporting TREM1 as an important receptor that participates in augmenting proinflammatory responses, and a null allele of TREM1 in mice results in diminished inflammatory reactions (Colonna 2003; Gibot and others 2007; Hommes and others 2014; Lagler and others 2009; Sharif and Knapp 2008). TREM1 signaling via TYROBP phosphorylation regulates NF-κB activation and expression of inflammatory genes (Arts and others 2012). TREM1 amplifies the innate immune response in part through synergistic crosstalk with pattern recognition receptors such as TLRs, which facilitate the response to a diverse range of pathogenic and inflammatory stimuli (Bleharski and others 2003; Dower and others 2008). In this way, TREM1 has been shown to play a regulatory role in influencing inflammatory disease outcomes, likely through its known role in activating antigen-presenting cells (APCs) and promoting their survival through several anti-apoptotic mechanisms (Cai and others 2013; Numata and others 2011; Prieto and others 2010; Yuan and others 2014). TREM1 may exert protective effects in AD by augmenting microglial activation and phagocytosis, as well as enhance microglial fitness through its antiapoptotic effects. TREM1 expression is decreased in the context of the rs6910730 risk variant, potentially impairing such effects and conferring AD risk (Chan and others 2015; Replogle and others 2015). However, few human studies have examined this topic, and the role of TREM1 in AD pathogenesis remains unclear.
SPI1
The SPI1 SNP rs1057233 has been established as a genetic risk factor for AD through GWAS. SPI1 encodes the protein PU.1, a well-known hematopoietic transcription factor that controls the expression of hundreds of myeloid genes (Huang and others 2017; Kunkle and others 2019). The SPI1 AD risk variant increases PU.1 expression (Huang and others 2017). PU.1 is involved in the early stages of hematopoietic progenitor cell development and in the later stages of myeloid cell maturation (Friedman 2007). Granulocyte-macrophage progenitor cells from mice deficient in PU.1 have impaired commitment to a macrophage fate, demonstrating that PU.1 tightly regulates monocyte and macrophage differentiation (Dakic and others 2005). Furthermore, PU.1 appears to be a nonclassical pioneer transcription factor, meaning it can reprogram cell-type fates (Minderjahn and others 2020). This capability is likely due in part to its ability to bind the chromatin remodeling complex SWI/SNF.
PU.1 regulates the activation and fitness of macrophages and monocytes, in part by regulating the expression of innate immune pattern recognition receptors including TLR1, TLR2, and TLR4. PU.1 binds elements within the proximal human TLR4 promoter and directly regulates its expression (Rehli and others 2000). In human monocytes, GM-CSF was found to downregulate TLR1, TLR2, and TLR4 in a PU.1-dependent manner (Sadeghi and others 2016). Reduced expression of these receptors resulted in decreased downstream p38 and ERK signaling and impaired proinflammatory cytokine production following exposure to lipopolysaccharide or lipoteichoic acid (Sadeghi and others 2016). In these ways, PU.1 acts as a critical regulator of innate immune responses to environmental triggers.
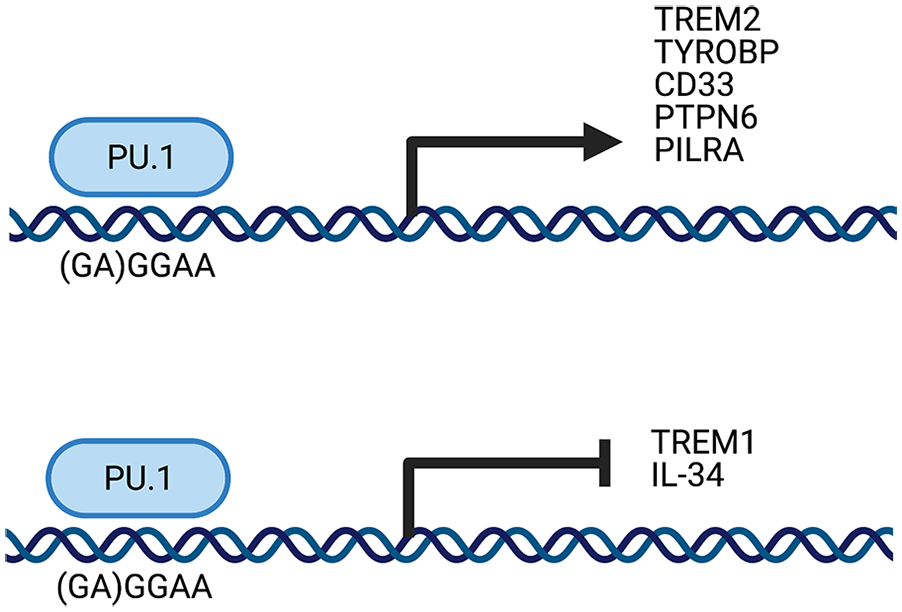
Interestingly, PU.1 also regulates the expression of several LOAD-associated genes (Fig. 1). This comes as little surprise, as PU.1 regulates many myeloid genes, and many LOAD-risk associated variants are enriched in myeloid cells. An overwhelming 60 genes that both lie in LOAD-associated loci and are expressed in microglia contain PU.1 binding sites (Huang and others 2017). Specifically, PU.1 regulates TREM2 expression, evidenced by studies in which silencing of SPI1 with siRNA led to reduced TREM2 expression (Rustenhoven and others 2018). PU.1 has also been established as a negative regulator of TREM1 (Zeng and others 2007). As the SPI1 risk allele increases PU.1 expression, the risk allele is associated with decreased TREM1 expression (Huang and others 2017). PU.1 also regulates TYROBP, through which TREM1 and TREM2 both signal, solidifying its regulation of the TREM1/TREM2/TYROBP axis at several points (Huang and others 2017). CD33 expression is also regulated by PU.1, and PU.1 is necessary for CD33 expression in myeloid cells (Bodger and Hart 1998). PU.1 additionally modulates PTPN6, the gene which encodes the protein SHP-1 (Wlodarski and others 2007). SHP-1 is the major downstream signaling molecule of CD33, demonstrating the importance of PU.1 in regulating several points in the CD33 signaling cascade (Taylor and others 1999). The PILRA gene also contains a PU.1 binding site, and PU.1 positively regulates its expression (Huang and others 2017). These findings clarify that PU.1 presents a point of convergence for regulating several LOAD genetic risk factors and their associated signaling cascades. Accordingly, PU.1 may serve as an advantageous therapeutic target for LOAD.
The LOAD genetically associated transcription factor PU.1 modulates the expression of other genetically associated genes (TREM2, CD33, PILRA, TREM1, and IL34) and their key signaling partners.
PLCG2
PLCG2, which encodes an enzyme that catalyzes the hydrolysis of phospholipids, was found to have a rare protective variant that is genetically associated with LOAD (Sims and others 2017). An independent association of PLCG2 with LOAD was also identified in a GWAS by proxy (Marioni and others 2018). A recent preprint manuscript confirmed the protective variant and the GWAS by proxy association in the promoter region, as well as identified an additional independent association with LOAD (de Rojas and others 2020). The rare AD-protective variant leads to an amino acid change, proline 522 to an arginine (P522R). This alteration increases PLCγ2 function, and knockin mice carrying the P522R mutation exhibit an enhanced acute inflammatory response (Magno and others 2019; Takalo and others 2020). This direction of effect is consistent with those of other AD genetic variants in which decreased microglial activation is associated with disease risk, and activation is associated with protection.
Interestingly, there are other known hypermorphic mutations in PLCG2 that are associated with autoinflammatory disease (Zhou and others 2012). The AD-protective variant has no such association with autoinflammation; increased PLCγ2 activity in the context of this variant is protective for AD neurodegeneration and supportive of longevity (van der Lee and others 2019). A more comprehensive understanding of these mutations is needed to differentiate detrimental and protective effects of increased PLCγ2 signaling in disease risk. It is possible that the mutations preferentially influence PLCγ2 participation in different signaling pathways, or that they disrupt interactions with regulatory binding partners that are differentially expressed in the various innate immune cells. Differential regulation of signaling pathways is possible, as the S707Y autoinflammatory mutation is located in the c-terminal SH domain, while the AD P522R mutation is located between the catalytic phosphatidylinositol-specific phospholipase C X domain and the regulatory N-terminal SH2 domain. Similarly, these mutations might differentially disrupt interactions between various domains and disturb autoinhibition.
PLCγ2 is a downstream signaling molecule of TREM2, TREM1, and several TLRs (Andreone and others 2020; Arts and others 2012; Bae and others 2017; Chae and others 2015; Chiang and others 2012; Zhu and others 2018). Its participation in TLR and TREM signaling pathways likely occurs through distinct mechanisms, as its ability to induce proinflammatory responses downstream of TLR2 signaling occurs through a TREM2 independent mechanism (Andreone and others 2020). In iPSC-derived microglia, PLCγ2 was shown to mediate TREM2 regulation of cholesterol metabolism (Andreone and others 2020). Further understanding of these genetic associations will clarify the role of PLCγ2 in AD and whether it may be modulated as a potential therapeutic target (de Rojas and others 2020).
IL34
An IL34 variant resulting in an early stop codon (Tyr213Ter) has been genetically associated with increased risk for AD (de Rojas and others 2020). IL-34 is produced by neurons, and may act as a neuronal signal to microglia (Mizuno and others 2011). IL-34 signals through the CSF1R, as does colony stimulating factor 1 (CSF-1), although IL-34 binds CSF1R with a higher affinity than CSF-1 (Lin and others 2008; Liu and others 2012). Both IL-34 and CSF-1 induce proliferation and cell survival in monocytes and other myeloid cells (Chitu and Stanley 2006; Dai and others 2002; Lin and others 2008; Wei and others 2010). While knockout of CSF-1 did not affect microglial development in mice, knockout of CSF1R resulted in microglial depletion (Wang and others 2012). These findings suggest that CSF1R-mediated signaling is essential for microglial development independent of CSF-1, with IL-34 potentially mediating this effect.
Interestingly, IL34 mRNA was found to be decreased in postmortem inferior temporal gyrus samples from AD patients compared to samples from MCI and controls, whereas CSF1R and CSF1 mRNA were both found to be increased (Walker and others 2017). This finding is consistent with the genetically associated variant resulting in an early stop codon, suggesting that decreased IL-34 expression is associated with AD risk. Of note, IL-34 and CSF1R functionally converge with other AD genetically associated molecules. Both IL-34 and CSF1R expression were found to be regulated by SPI1/PU.1 in BV2 cells (Huang and others 2017). Additionally, CSF1R interacts with PLCγ2 as a binding partner (Bourette and others 1997). Treatment with IL-34 was found to promote microglial proliferation and uptake of soluble oligomeric Aβ in vitro, as well as to reduce neurotoxic effects of Aβ (Ma and others 2012). Additionally, IL-34 signaling decreases oxidative stress and facilitates neuronal survival (Luo and others 2013; Ma and others 2012). IL-34 may similarly provide cognitive benefits, as intracerebral administration of IL-34 reduced impairments in associative learning in AD mice (Mizuno and others 2011). These findings further support the protective effects of IL-34 in AD, as well as the AD genetic risk associated with the truncating Tyr213Ter mutation.
Despite these protective effects of IL-34 signaling via CSF1R, a clinical trial is currently underway for a CSF1R antagonist as a therapeutic approach for AD, to reduce the microglial population as well as to suppress their activity (Raymont 2020). As decreased IL-34 signaling via CSF1R is associated with AD risk as reviewed above, it may instead be therapeutically advantageous to activate CSF1R in AD. However, ablation of an immune cell type has been found to be a beneficial therapeutic approach in some autoimmune diseases, such as the use of B cell depletion therapy in the treatment of multiple sclerosis (Greenfield and Hauser 2018).
HLA
Somewhat surprisingly, the human leukocyte antigen (HLA) complex region is another genetically associated locus that implicates the innate immune system in LOAD pathogenesis (Lambert and others 2013). HLA is one of the strongest genetic associations for most autoimmune diseases, in which the adaptive immune system engages in dysregulated, self-reactive responses. HLA variants have now been identified as genetic risk factors for several neurodegenerative diseases (Barcellos and others 2006; Hamza and others 2010; Jansen and others 2019; Lambert and others 2009; Nalls and others 2014; Steele and others 2017; Wissemann and others 2013). Genetic variants in both major histocompatibility complex (MHC) class I and class II have been associated with LOAD (Steele and others 2017). MHC class II is almost exclusively expressed on innate immune cells, and therefore is predominantly expressed on microglia within the CNS. MHC class I has also been identified as a microglial enriched gene in brains from aged individuals, although it can be expressed by all cell types (Olah and others 2018). Antigen-presenting cells (APCs) including microglia, utilize MHC class II molecules (HLA-DR, HLA-DQ, HLA-DP) to present antigen to CD4+ helper T cells. Similarly, cells utilize MHC class I molecules (HLA-A, HLA-B, HLA-C) to present antigen to CD8+ cytotoxic T cells.
Both an MHC class I haplotype ((A*03:01~B*07:02) and a class II haplotype (DRB1*15:01- DQA1*01:02- DQB1*06:02 (DR15)) were found to be genetically associated with LOAD (Kunkle and others 2019; Steele and others 2017). This same class II haplotype, DR15, has also been associated with Parkinson’s disease and multiple sclerosis (Barcellos and others 2006; Wissemann and others 2013). Different HLA molecules have varying specificities with which they bind antigen peptides. The association of DR15 with autoimmune disease suggests its proclivity in presenting self-peptides, in some circumstances initiating a self-reactive adaptive immune response. It has also been suggested that a large hydrophobic pocket in DR15’s peptide-binding groove allows for metabolite binding in addition to peptides (Misra and others 2019). These small molecules are abundant and are often derived from the human microbiome. NAD+ is one such metabolite that is predicted to bind this pocket (Misra and others 2019). A number of studies, including mouse model studies of AD, have established decreased NAD+ as a hallmark of aging and have linked its decrease to the development of chronic diseases (Braidy and others 2018; Demarest and others 2019; Lautrup and others 2019; Schultz and Sinclair 2016).
These HLA genetic findings solidify the involvement of innate immune cells, presumably microglia, in LOAD susceptibility. They also suggest an important role for the interface between the innate and adaptive immune systems in disease pathogenesis and a role for microglia as APCs. This microglial T cell interface may be especially important in the hippocampus, where extravascular CD4+ and CD8+ T cells have been found to be increased in LOAD (Itagaki and others 1988; Merlini and others 2018; Rogers and others 1988). It has been reported that there are more CD8+ T cells than CD4+ T cells among the LOAD infiltrating T cells (Merlini and others 2018). However, no detailed characterization of the T cell phenotype has yet been performed. Future work may reveal the nature of their interaction with microglia and their role in brain neuropathology.
Convergence of GWAS Loci and Functional Outcomes
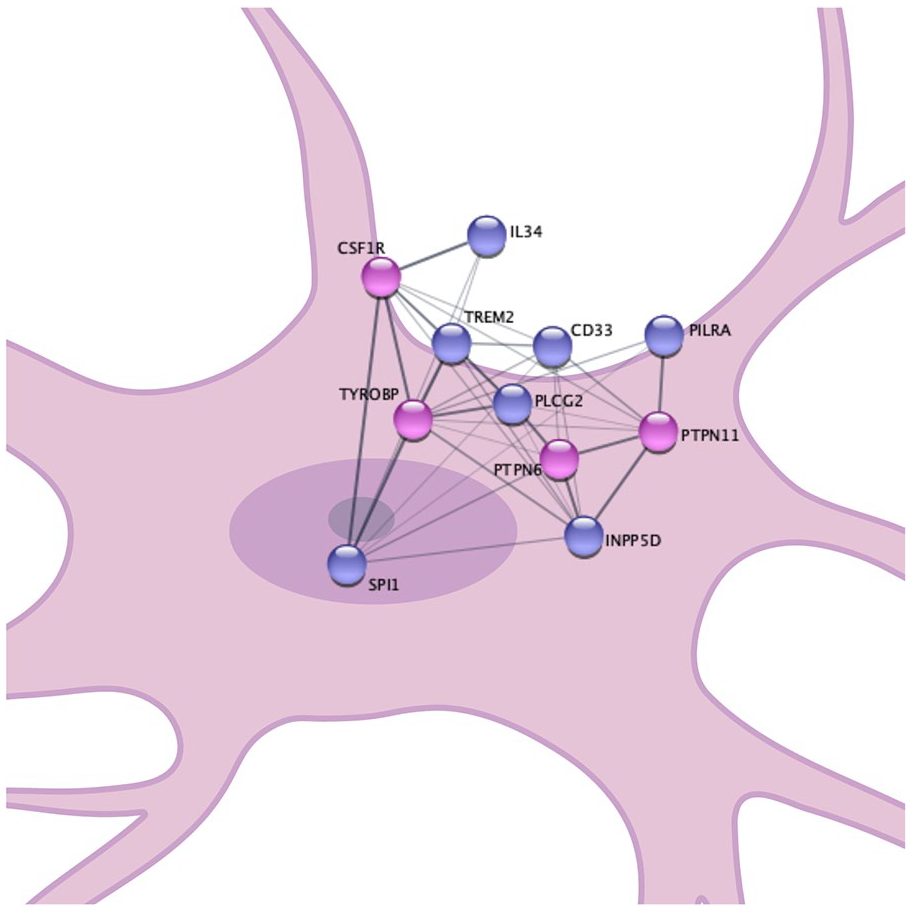
Many of the proteins genetically associated with LOAD reviewed here appear to interact and share pathways or regulatory mechanisms (Fig. 2). These findings present several points of convergence that may be strategic targets for therapeutic intervention. In summary, CD33 functions upstream of TREM2 and regulates TREM2 expression (Chan and others 2015; Griciuc and others 2019). The TREM2/TREM1/TYROBP axis described in this review is tightly interconnected and appears to be modulated by PU.1 (Arts and others 2012; Bouchon and others 2001; Chan and others 2015; Huang and others 2017). PLCγ2 functions downstream of TREM1, TREM2, and TYROBP, providing another point in this axis that may be advantageous to manipulate therapeutically (Andreone and others 2020; Arts and others 2012). PU.1 also regulates CD33, PILRA, IL-34, and CSF1R (the IL-34 receptor). Interestingly, the SPI1 AD risk variant appears to increase expression of CD33 and PILRA and to decrease expression of TREM1 and IL-34. These outcomes are consistent with the alterations in protein expression or function observed with the CD33, PILRA, TREM1, and IL34 risk variants, further supporting the association of these patterns of protein expression with LOAD risk. PU.1 may therefore present a particularly advantageous therapeutic target as it modulates the expression of several genetically associated LOAD risk genes.
Many of the proteins that are coded for by LOAD-associated genes interact with each other and are in shared microglial signaling pathways; proteins in dark purple are encoded by genes in LOAD genetic loci, proteins in light purple are critical binding partners.
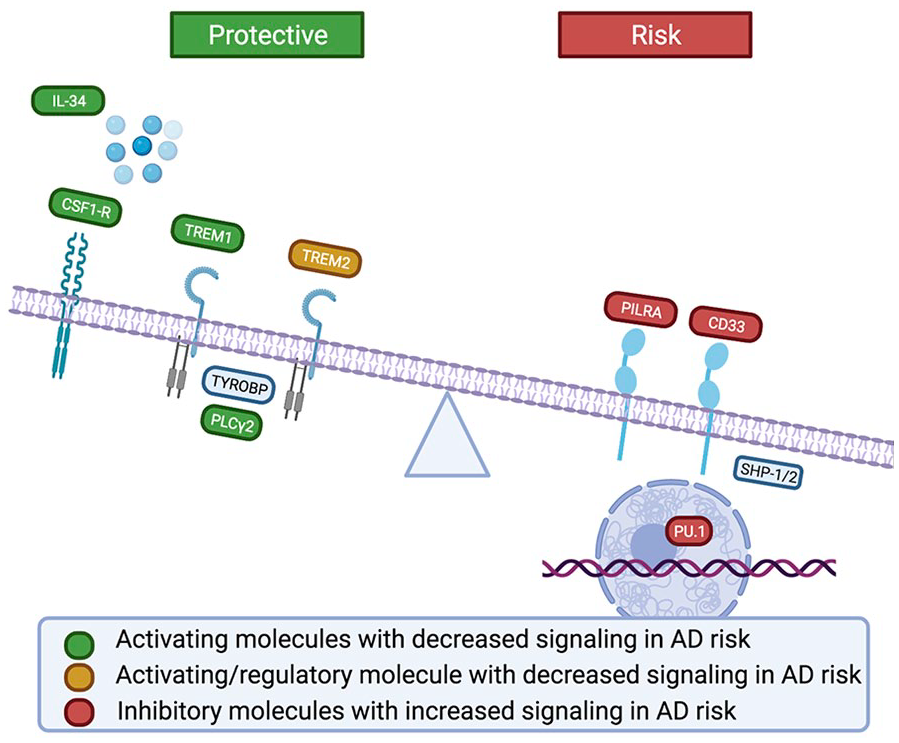
In addition to the convergence of molecular pathways, it is valuable to consider whether these risk variants converge on a shared microglial functional outcome that may contribute to AD pathogenesis. Of the reviewed genetically identified variants enriched in microglia for which a direction of effect has been established, the findings suggest that a less responsive microglial phenotype is associated with AD risk. Supporting this trend, the risk alleles in CD33, TREM1, TREM2, PILRA, SPI1, and IL34 converge to suppress immune signaling, while the protective allele in PLCG2 enhances the immune response (Fig. 3; Bradshaw and others 2013; de Rojas and others 2020; Huang and others 2017; Kleinberger and others 2014; Magno and others 2019; Rathore and others 2018; Replogle and others 2015). These findings are in stark contrast to the dogma that arose in the pre-GWAS era that concluded that the immune system needed to be suppressed (Akiyama and others 2000).
AD genetic risk is associated with decreased signaling of activating microglial receptors, and increased signaling of inhibitory receptors, resulting in a dampened microglial response in AD.
The AD brain environment prominently features sterile inflammatory stimuli, such as highly inflammatory molecules released by deteriorating or dead cells and misfolded protein aggregates. Microglial suppression resulting in the inability to effectively resolve these sterile inflammatory processes, mitigate pathology, and maintain brain homeostasis may heighten susceptibility to neuronal injury. Indeed, the inability to clear pathology is thought to be critical to AD progression (Fiala and others 2005; Fiala and others 2007; Hickman and others 2008; Wildsmith and others 2013; Yoon and Jo 2012). This phenotype may be mirrored by senescent deterioration associated with aging, the primary risk factor for Alzheimer’s disease (Damani and others 2011; Hefendehl and others 2014; Kronenberg and others 2017; Mosher and Wyss-Coray 2014; Streit 2006; Streit and others 2004). Therefore, the genetic findings discussed in this review support the loss of microglial fitness in AD, and may converge with the functional outcomes of other known LOAD risk factors on an ineffective microglial phenotype. A number of ongoing clinical trials, as mentioned above, will shed more light on this topic.
Genetics, Microglial Immune Responses, and the Pathogen Hypothesis
In addition to the amyloid cascade and tau hypotheses, it has been suggested that LOAD pathogenesis may be triggered by an infectious agent, a controversial and debated concept referred to as the “pathogen hypothesis” (Li and others 2018). Viral, fungal, and bacterial pathogens have been associated with LOAD, including herpes simplex virus type 1 (HSV-1), human herpesvirus 6A (HHV-6A), and HHV-7, as well as the intracellular bacteria Chlamydophila pneumoniae and
In human subjects, LOAD genetic risk variants appear to converge to dampen the innate immune response, as detailed in this review. Microglial enrichment for these risk variants is suggestive of a suppressed microglial phenotype as a pathological mechanism of disease. Importantly, age is the number one risk factor for LOAD. Age also profoundly impacts the immune system’s fitness, similarly converging with the functional outcomes of the reviewed LOAD genetic risk variants to suppress immune function (Del Giudice and others 2018). An aged immune system, as well as carriage of suppressive LOAD genetic variants, may alter the local microglial response to infectious and sterile inflammatory stressors. Some AD risk variants may also directly alter host-pathogen interactions. For example, glycoprotein B of herpes simplex virus 1 (HSV-1) has been identified as a ligand for PILRA (Satoh and others 2008). As the AD-protective variant leads to reduced PILRA ligand binding, viral entry into the host cell via glycoprotein B may be limited (Rathore and others 2018). The HLA DR15 association has been shown to lead to a higher viral load and attenuated immune control of EBV in a model of multiple sclerosis (Zdimerova and others 2021). Additionally, the HLA genetic associations with LOAD raise the question of what antigens are potentially being presented by microglia to CNS-infiltrating T cells in AD. Are these self-antigens modified by the disease process such as misfolded proteins, or peptides from pathogens that are residing in the CNS? Further study of microglial-T cell interactions will lead to a better understanding of the inflammatory stimuli present in the CNS that may be contributing to LOAD, and the immune system’s ability to respond to these challenges.
Rather than an active infection contributing to LOAD, it is possible that infectious processes over a lifetime may act as triggering events in AD pathogenesis, or exacerbate preexisting disease processes. Inflammatory challenges, both sterile and infectious, are known to result in long term alteration of immune dynamics and may facilitate immunosenescence (Coppé and others 2010; Dregan and others 2014; Effros 2016; Fairweather and others 2012; Furman and others 2019; Jose and others 2017; Pawelec and others 2005; Root-Bernstein and Fairweather 2014; Stack and others 2012). An inflammatory insult may prime microglial activation or dysfunction, dictated by factors including genetic susceptibility and the nature of the challenge. These challenges may alter functional programs of microglia, which may be potentiated long after the resolution of an infection or sterile inflammatory process. In this way, a lifetime burden of inflammatory challenges may alter microglial fitness and function. Such alterations, compounded in the context of immune aging and suppressive innate immune genetic risk variants, may contribute to disease. Importantly, inflammatory challenges need not be restricted locally within the CNS. Systemic inflammatory challenges have been shown to result in sustained alterations of microglial functional programs in mice (Wendeln and others 2018). In humans, systemic infections have been demonstrated to modify the late-stage immune response in AD, resulting in an immunosuppressive brain environment, a finding that converges with the impaired immune environment associated with AD risk as outlined in this review (Rakic and others 2018).
Concluding Thoughts
The genetic variants described in this review implicate a role for suppressed innate immune function, particularly in microglia, in LOAD pathogenesis. Innate immune dysregulation likely results in microglial inability to mitigate pathologies and resolve inflammatory processes, whether sterile or pathogenic, in AD. In the context of age and the accompanying immunosenescence, as well as immune alterations resulting from pathogenic and sterile inflammatory challenges, a dampened microglial phenotype may give way to microglial failure. This microglial failure may lead to a loss of homeostasis in the local brain environment and increased vulnerability to neuronal injury and toxicity. As AD is a progressive disease with age as its primary risk factor, and as microglia live on average for 4 years and up to two decades, it is particularly important to consider the dynamics of increased suppressive tone in microglia over several years of inflammatory challenges (Réu and others 2017). Future research must uncover the consequences of the dampened immune tone resulting from these genetic variants on microglial function, and how impairments in microglial capability participate in LOAD pathogenesis.
Footnotes
Acknowledgements
Figures 2 and 3 and Box 2 were created in BioRender. Figure 1 was made using String, Cytoscape, and BioRender.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: EMB is a founder of IMAD Therapeutics. ZKC has no conflicts of interest to disclose.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.