
Abstract
Windows of plasticity are fundamental for the correct formation of definitive brain circuits; these periods drive sensory and motor learning during development and ultimately learning and memory in adults. However, establishing windows of plasticity also imposes limitations on the central nervous system in terms of its capacity to recover from injury. Recent evidence highlights the important role that astrocytes and adenosine seem to play in controlling the duration of these critical periods of plasticity.
Importance of Windows of Plasticity
It is known that the ability to play a musical instrument, to learn a language, or to develop sensory and motor skills is associated with periods of plasticity during development. Indeed, it is generally assumed that these skills are better acquired when trained at younger ages, which is attributed to the plasticity in the brain during specific periods of pre- and postnatal development. Plasticity is the ability to induce modifications in response to experience or external stimuli, and it is one of the most interesting properties of the mammalian brain. Indeed, plasticity is involved in the organization of cortical maps during development, as well as in learning and memory processes in the adult, and the disruption of plasticity in early postnatal periods may underlie neurodevelopmental diseases. Since it was first discovered (Bliss and others 2013; Cajal 1984; Mateos-Aparicio and Rodríguez-Moreno 2019), it was clear that synaptic plasticity is a phenomenon that varies over the individual’s lifetime and that it is a changing and evolving process. Indeed, critical and permissive periods of plasticity exist during development that are referred to as windows of plasticity. Intense activity/sensory-dependent plasticity occurs in these windows, with environmental experiences exerting the strongest impact on brain circuits in the permissive periods and with these inputs driving maximal plasticity in specific brain circuits during the critical periods. The reordering and refinement of neural connections in these windows of plasticity establish the definitive synapses and circuits responsible for the correct physiology in adults (Hensch 2004). After these critical periods, there is a loss of plasticity at particular synapses (Hensch 2004, 2005). Accordingly, there has been much interest in better understanding the mechanisms and secrets behind the critical periods of plasticity in recent years, not least with a view to possibly develop new therapies for pathologic conditions in humans (Dromerick and others 2021) and to correct miswired neuronal circuits. Moreover, such information may help improve educational training and rehabilitation, enhancing recovery from traumatic injury and lesions in the adult brain. One of the fundamental questions in the field is whether the windows of critical periods can be modified or recovered later in life. Significantly, the answer to this question seems to be “yes,” and what is more, GABAergic activity and adenosine and astrocyte signaling seem to be fundamental elements in these processes.
Influence of GABAergic Inhibition on the Windows of Plasticity
There is increasing evidence that GABAergic inhibition is crucial for certain aspects of brain development and to control certain windows of plasticity. Since the experiments of Hubel and Wiesel (2004) >55 y ago and those of Hensch (2005) on amblyopia, GABAergic inhibition has been proposed as a mechanism to control critical periods of plasticity (Hensch 2004). This is true at some synapses in the somatosensory and visual cortices where GABAergic inhibition was seen to be involved in these periods of plasticity (Hensch 2004), yet it is not clear whether this is a general mechanism that acts in all brain regions. The evidence available seems to suggest that this is not the case, as it was recently seen that astrocytes and adenosine fulfill a crucial role in controlling or defining the duration of the critical period of plasticity in the auditory and somatosensory cortices and in the hippocampus, processes in which GABAergic inhibition is not involved (Blundon and others 2017; Falcón-Moya and others 2020; Martínez-Gallego, Pérez-Rodríguez, and others 2022; Pérez-Rodríguez and others 2019). Here we consider the importance of astrocyte and adenosine signaling in controlling the onset and duration of critical periods of neuronal plasticity.
Astrocytes and Adenosine Influence the Dynamics of Plasticity during Postnatal Development
Recent evidence indicates that events unrelated to GABAergic inhibition may control critical periods of plasticity. Thus, at CA3–CA1 synapses in the hippocampus, a form of presynaptic spike timing-dependent long-term depression (t-LTD) was recently discovered that is present during the first weeks of mouse development but disappears during the fourth week of postnatal life (Andrade-Talavera and others 2016; Falcón-Moya and others 2020; Martínez-Gallego, Pérez-Rodríguez, and others 2022; Pérez-Rodríguez and others 2019). The mechanisms involved in this loss of plasticity include enhanced adenosine activation of presynaptic type 1 adenosine receptors (A1Rs) with maturation. Interestingly, the adenosine that activates A1Rs is most probably released by astrocytes, as the effects of this A1Rs activation are always observed just after astrocyte stimulation or signaling is induced (Figure 1). Indeed, it was elegantly demonstrated that an antagonist of A1R recovers t-LTD when it is lost after the fourth week of development. Moreover, in the presence of an agonist of A1R, the window of plasticity can be closed and can be t-LTD impaired during the first 3 wk of postnatal development, shortening of this critical period of plasticity. This is a clear indication of how the duration of the critical period of plasticity can be manipulated pharmacologically during postnatal development. However, it remains to be determined whether the adenosine that mediates this loss of plasticity is directly or indirectly released by astrocytes. It is particularly notable that once the critical period of t-LTD at CA3–CA1 synapses has terminated during the fourth week of postnatal development, the same protocol of plasticity induces timing-dependent long-term potentiation (t-LTP) instead. Thus, the natural series of events seems to be the existence of t-LTD for 3 to 4 wk, followed by a switch from t-LTD to t-LTP in response to the same (natural) stimuli (Falcón-Moya and others 2020). This switch from t-LTD to t-LTP with maturation is also mediated by the increased levels of adenosine and enhanced A1R activation that persist after the critical period for t-LTD closes. Moreover, this increase in adenosine continues to be dependent on astrocyte signaling, as the switch requires astrocyte gliotransmitter release, reflecting the central role of astrocytes in controlling the duration of these periods of plasticity (Falcón-Moya and others 2020; Figure 1).
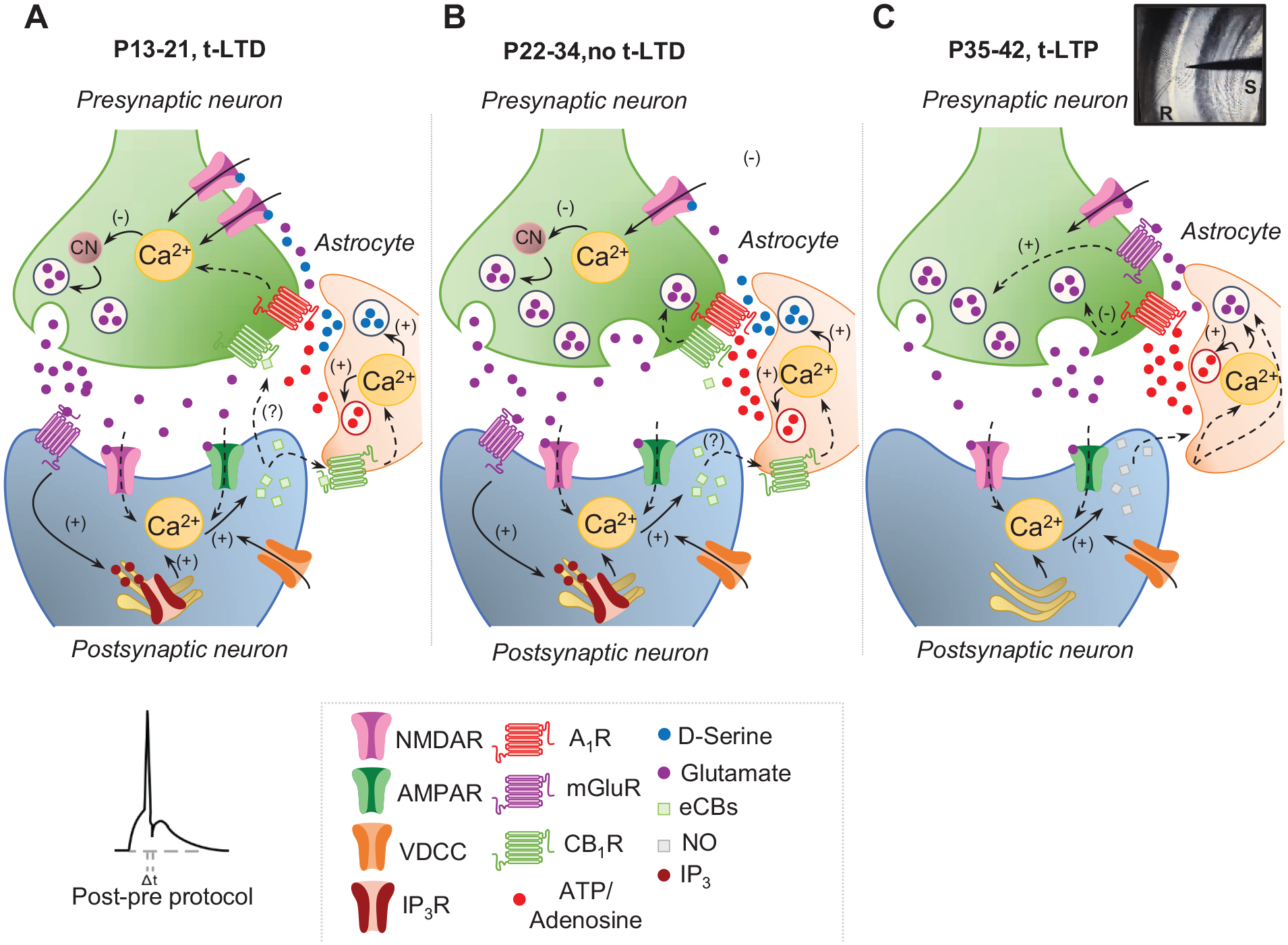
Scheme showing signaling at early (P13–P21), juvenile (P22–P34), and adult (P35–P42) stages of development at CA3–CA1 synapses of the hippocampus. (A) At P13 to P21, a presynaptic form of t-LTD is induced by a post-pre single-spike pairing protocol (i.e., postsynaptic action potential preceded presynaptic stimulation). In this form of t-LTD, postsynaptic action potentials activate VDCCs, and the presynaptically released glutamate activates postsynaptic mGluR, activating PLC and provoking Ca2+ release from internal stores and DAG production, which serves as a precursor for eCB synthesis. For t-LTD, an eCB signal is necessary to activate CB1 receptors to facilitate
Very recently, a similar mechanism for t-LTD was observed at synapses of the somatosensory cortex from layer 4 to layer 2/3 (L4–L2/3); specifically, there is a form of t-LTD at L4–L2/3 synapses in the somatosensory cortex that disappears after the first weeks of postnatal development (in this case during the fifth week). Here, as in the hippocampus, the loss of plasticity is due to an increase in the activation of presynaptic A1Rs and astrocytic signaling (Martínez-Gallego, Pérez-Rodríguez, and others 2022). Results are consistent with the predictions of biophysicochemical models of somatosensory cortical L4–L2/3 t-LTD in vivo (Linne and others 2022; Manninen and others 2020). In addition, and as observed in the hippocampus (Falcón-Moya and others 2020), a switch from t-LTD to t-LTP is seen once t-LTD is lost, again because of the continued increase in adenosine and astrocyte signaling with maturation (Martínez-Gallego, Pérez-Rodríguez, and others 2022; Figure 2). The fact that a mechanism involving adenosine and astrocyte signaling controls the onset and duration of a critical period of plasticity suggests that this may be a general mechanism to control the duration of critical periods of plasticity in the central nervous system, probably because of their effect on presynaptic glutamate release and the differential activation of N-methyl-
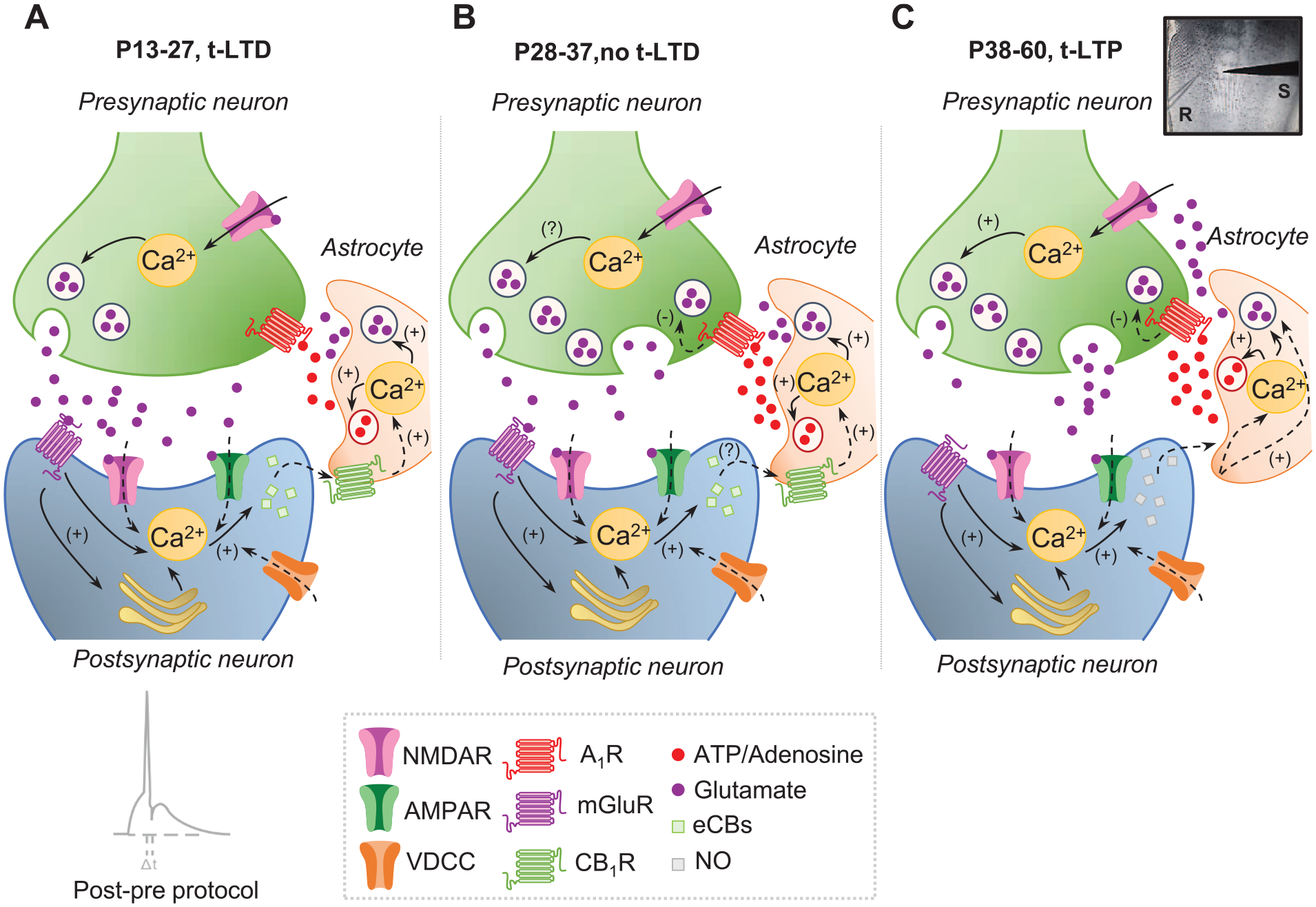
Scheme showing signaling at early (P13–P27), juvenile (P28–P37), and adult (P38–P60) stages of development at synapses of the somatosensory cortex from layer 4 to layer 2/3 (L4–L2/3). (A) At P13 to P27, a presynaptic form of t-LTD is induced by a post-pre protocol (i.e., postsynaptic action potential preceded presynaptic stimulation). In this t-LTD, postsynaptic action potentials activate VDCCs, and the presynaptically released glutamate activates postsynaptic mGluR, provoking Ca2+ release from internal stores and eCB synthesis. For t-LTD, an eCB signal is necessary to activate CB1 receptors to facilitate glutamate release from astrocytes. With the glutamate released from presynaptic neurons, this glutamate is known to activate presynaptic NMDARs on L4 boutons, leading to an increase in presynaptic Ca2+ release and synaptic depression. (B) At P28 to P37, t-LTD does not develop, and the main difference with regard to P13 to P27 is an increase in adenosine release from astrocytes at P28 to P37 as compared with P13 to P27. (C) At P38 to P60, t-LTD is not only lost but also switched to a presynaptic form of t-LTP. For the induction of this presynaptic form of t-LTP, postsynaptic action potentials activate VDCCs, causing calcium release from internal stores and inducing NO synthesis. The NO signal leads to the activation of astrocytes to release glutamate and/or adenosine to activate NMDARs and A1R, respectively, on L4 boutons. A1R activation considerably reduces neurotransmitter probability release, whereas NMDAR activation leads to a long-lasting increase in glutamate release and synaptic potentiation. Modified from Martínez-Gallego, Pérez-Rodríguez, and others (2022). A1R = type 1 adenosine receptor; AMPAR = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; CB1R = type 1 cannabinoid receptor; eCB = endocannabinoid; mGluR = metabotropic glutamate receptor; NMDAR = N-methyl-
Interesting discoveries have also been made at thalamocortical synapses. Thalamocortical LTD or LTP cannot normally be induced in slices from animals older than 2 to 3 wk of age, and LTP and LTD at thalamocortical projections are known to be restricted to the same critical period as cortical map plasticity (Crair and Malenka 1995). Significantly, a form of LTD not present in adults is rescued when adenosine is downregulated by the activation of presynaptic muscarinic M1 receptors in brain slices containing the auditory thalamus and cortex (Blundon and others 2011). Hence, the release of presynaptic gating mechanisms appears to allow mature thalamocortical synapses to undergo LTD. Interestingly, GABAergic synapses do not appear to participate in this process, as LTD is present in the presence of picrotoxin (Blundon and others 2011). At the same synapses, LTP can be induced in young animals but is not normally observed at postnatal days 35 to 42. This form of LTP is also gated by adenosine from the thalamus in adults as it can be rescued by dampening presynaptic A1R activation (Chun and others 2013). It is noteworthy that experiments in vivo showed that windows of thalamocortical synapse plasticity can be closed by activating A1Rs in young animals (Blundon and others 2017). In addition, blocking or deleting A1Rs at postnatal days 45 to 56 improves cortical map plasticity, and it recovers LTP and LTD beyond the critical period. However, the possible role of astrocytes that release ATP/adenosine at thalamocortical auditory synapses has yet to be explored, as is also the case for the role of adenosine in vivo in the hippocampus and somatosensory cortex.
As indicated, GABAergic and adenosine signaling appears to influence the duration of critical periods of plasticity. Adenosine (released directly or as ATP that is then converted to adenosine in the extracellular medium) seems to be released directly by astrocytes in the hippocampus (Pérez-Rodríguez and others 2019) and somatosensory cortex (Martínez-Gallego, Pérez-Rodríguez, and others 2022), and in these two regions astrocyte signaling has clearly been established as being necessary for the loss of t-LTD. Nevertheless, it remains possible that adenosine is released indirectly after astrocyte gliotransmitter release, which will need to be unequivocally clarified by further research. Apart from the earlier studies that first described a role for astrocytes in closing a critical period of plasticity (Falcón-Moya and others 2020; Pérez-Rodríguez and others 2019), astrocytes were recently found to control the closure of a critical window in the mouse visual cortex by affecting extracellular matrix proteins (Ribot and others 2021). Likewise, in Drosophila melanogaster, astrocytes were seen to control the closing of a critical period of plasticity in a motor circuit by affecting neuroligin and neurexin signaling (Ackerman and others 2021). Whether astrocytes participate in the release of adenosine in these two critical periods remains to be determined.
Conclusions
In addition to other mechanisms, recent evidence indicates that astrocytes and adenosine are new and important elements that can define the duration of critical periods of plasticity. Combinations of sensory motor training and pharmacologic or molecular interventions may act synergistically to enhance plasticity and permit functionality to be recovered after injury. The possible pharmacologic manipulations mentioned here may also have transcendental consequences, allowing closed windows of plasticity to be opened and permit renewed learning, which may pave the way to combat some neurologic diseases. As such, these are currently exciting times for those interested in understanding how, and through what natural stimuli, critical periods of plasticity are controlled.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research in the Rodríguez-Moreno laboratory has been supported by the Agencia Estatal de Investigación/FEDER (grant PID2019-107677GB-I00) and Junta de Andalucía/FEDER (grant P20_0881).