
Abstract
The tau protein is a key contributor to multiple neurodegenerative diseases. The pathology of tau is thought to be related to tau’s propensity to form self-templating fibrillar structures that allow tau fibers to propagate in the brain by prion-like mechanisms. Unresolved issues with respect to tau pathology are how the normal function of tau and its misregulation contribute to disease, how cofactors and cellular organelles influence the initiation and propagation of tau fibers, and determining the mechanism of tau toxicity. Herein, we review the connection between tau and degenerative diseases, the basis for tau fibrilization, and how that process interacts with cellular molecules and organelles. One emerging theme is that tau interacts with RNA and RNA-binding proteins, normally and in pathologic aggregates, which may provide insight into alterations in RNA regulation observed in disease.
Keywords
Introduction
Intracellular aggregation and hyperphosphorylation of microtubule-associated protein tau, or tau, are implicated in a group of neurodegenerative diseases collectively referred to as tauopathies (Kovacs 2017). This group of diseases encompasses Alzheimer disease (AD), frontotemporal dementia (FTD), and chronic traumatic encephalopathy. The most prevalent, AD, currently affects >5.5 million Americans and is the sixth-leading cause of death in the United States (Heron 2019; Mayeux and Stern 2012). Owing to its long disease course and lack of treatment options, AD is also one of the most expensive diseases to treat, costing the US medical system an estimated $305 billion in 2020 (Alzheimer’s Association 2020). The number of people with AD and associated medical expenses are expected to triple by the year 2050 (Alzheimer’s Association 2020).
Emerging evidence suggests that tau interacts with RNA and RNA-binding proteins (RBPs) and that these interactions could be important for the formation and toxicity of tau aggregates. Herein, we discuss the mechanisms and implications of tau’s interactions with RNA and RBPs in health and disease. To place this discussion in context, we discuss tau’s ability to form fibrils that propagate in a prion-like manner, the evidence that tau aggregates display toxicity, and how RNA and RBPs are involved.
Tau has the capacity to form fibrils that self-template and spread in a prion-like manner (Dujardin and Hyman 2019; Figure 1). A unit capable of self-templated growth is referred to as a seed. Seeds can grow by inducing soluble monomeric tau to take on a similar fibrillar structure by stacking one on top of the other and creating fibrils stabilized by β-sheet and cross-β interactions (Mudher and others 2017). Seeds can be released from neurons during death or can cross the plasma membrane and be taken up by neighboring neurons, astrocytes, and microglia (Rauch and others 2020).
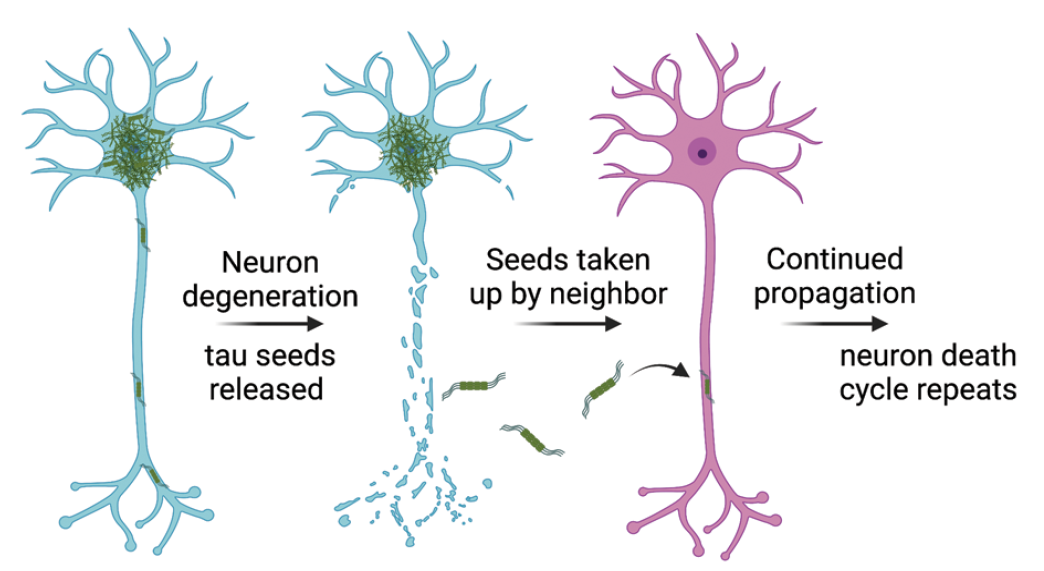
Prion-like propagation of tau aggregates. Tau has the capacity to spread in a prion-like manner whereby misfolded tau “seeds,” thought to be composed of tau oligomers, can be released from live neurons or neurons that have degenerated. Neighboring neurons, astrocytes, and microglia can acquire released tau seeds, which are thought to then template the conversion of soluble tau into more misfolded tau.
Several lines of evidence suggest that the formation and propagation of tau oligomers and/or aggregates are key drivers of toxicity in tauopathies. First, mutations in the MAPT gene that promote tau aggregation are causative of early-onset neurodegeneration in FTD with parkinsonism–17 (Goedert and Spillantini 2000). Second, cognitive decline in AD is closely related to tau aggregate deposition (Bierer and others 1995; Hanseeuw and others 2019; Riley and others 2002). Third, the reduction of tau is neuroprotective in mouse models of AD (DeVos and others 2018; SantaCruz and others 2005). This suggests that tau aggregation plays a critical role in neurodegeneration, yet a mechanistic link between tau and neurotoxicity has yet to be well defined.
It has been suggested that the smaller prefibrillar oligomeric tau aggregates are more toxic and display greater seeding propensity than the large fibrillar neurofibrillary tangles (NFTs) seen late in disease progression. NFTs make up the large hyperphosphorylated aggregates seen in the neurons of patients with tauopathies and are insoluble in a 1% sarkosyl detergent (Kuchibhotla and others 2014).
Oligomers, however, are generally defined as aberrantly folded tau multimers that are soluble in 1% sarkosyl, yet the precise methodology for isolating, generating, and identifying tau oligomers differs among research groups and can refer to a range of tau forms (Lasagna-Reeves and others 2012; Maeda and Takashima 2019; Shafiei and others 2017). Direct comparisons in cell culture models have shown that small oligomers are more toxic and promote more prion-like propagation than large fibrils (Ghag and others 2018; Mirbaha and others 2015; Zwierzchowski-Zarate and others 2022). Similarly, NFTs do not appear to disrupt neuronal functioning in awake mice (Kuchibhotla and others 2014). In contrast to tau fibrils, where the cryo–electron microscopy (cryo-EM) structures of several tauopathies have been solved, the precise structural conformation of tau oligomers and how they differ among tauopathies are poorly understood. As many histopathologic descriptions of tau aggregates in disease do not discriminate between oligomeric forms and larger fibrillar aggregates, we use the term tau aggregates as encompassing the tau fibrils and tau deposits seen microscopically.
Recent cryo-EM studies of the tau fibril structures isolated from postmortem samples of patients with a variety of tauopathies displayed a diversity of fibril forms (Falcon and others 2018; Fitzpatrick and others 2017; Katsumoto and others 2019; W. Zhang and others 2020). These fibril structures can differ in the exact region of tau that is involved, the amino acid modifications, and the nonproteinaceous cofactors that are observed to be associated with the fibrils. The identity of these cofactors in neurons remains to be determined. Polyanionic cofactors such as RNA and heparin promote the fibrilization of tau in vitro, yet these structures do not precisely match the fibrillar forms from patient brains (Abskharon and others 2022; Lövestam and others 2022; Zhang and others 2019).
Tau’s physiologic function is thought to be involved in regulating microtubules (Drubin and Kirschner 1986). However, several studies suggest that tau may function as an RBP and that interactions with RNA and other RBPs may contribute to tau’s toxicity. In fact, one of the earliest observations on tau showed that RNA binds to tau and inhibits its ability to promote the assembly of tubulin in vitro (Bryan and others 1975). This inhibition was stoichiometric, relieved by digestion of the RNA with RNase A, and sequence independent, as replicated via RNAs from sea urchins, CHO cells, and beef brain, as well as poly(A), (C), (G), and (U). This raises the possibility that tau might also function as an RBP.
Since these initial observations, several additional key pieces of data have pointed toward tau’s role as an RBP. First, tau has been shown to bind RNA in vitro and in vivo (Abskharon and others 2022; Fichou and others 2018; Zhang and others 2017). Second, RNA stimulates the formation of tau fibrils in vitro, and a cryo-EM structure of the resulting fibrils showed that RNA stably adhered to the tau fibril (Abskharon and others 2022; Dinkel and others 2015; Lövestam and others 2022). Third, RNA colocalizes with tau aggregates in mouse brains and the brains of patients with tauopathies (Ginsberg and others 1997; Ginsberg and others 1998; Lester and others 2021). Fourth, either the monomeric or aggregated forms of tau have been reported to localize to RNA-rich assemblies, including splicing speckles, the nucleolus, and stress granules (Lester and others 2021; Thurston and others 1997; Vanderweyde and others 2012), as well as associate with some RBPs and ribosomal proteins as assessed by coimmunoprecipitation (Gunawardana and others 2015; Koren and others 2019; Maziuk and others 2018; Tracy and others 2022). Finally, these associations of tau with RNA and RBPs appear relevant to disease since RNase treating tau aggregates isolated from the brains of patients with AD leads to tau fibril breakdown and a dramatic reduction in prion-like propagation of tau (Abskharon and others 2022; Zwierzchowski-Zarate and others 2022). Mutations in RBPs have also been found to be enhancers and suppressors of tau toxicity in Drosophila, Caenorhabditis elegans, and mouse models of tauopathy (Apicco and others 2018; Guthrie and others 2011; Shulman and Feany 2003; Wheeler and others 2019). Herein, we focus on tau and its potential physiologic and pathologic roles as an RBP.
Structure and Isoforms of the Tau Protein
To understand tau aggregation and function, one needs to consider the organization and isoforms of the tau protein. In the central nervous system, the full-length tau protein is composed of four main domains: an N-terminal projection domain, a proline-rich domain, a microtubule-binding domain (MTBD; the critical domain that can form self-propagating fibrillar structures), and a C-terminal domain. Alternative splicing exons 2 and 3 control the number of N terminal projection domains (0N, 1N, or 2N), and alternative splicing of exon 10 controls the number of repeats in the MTBD (either 3R or 4R; Figure 2). In total these splicing changes give rise to six isoforms in the human brain.
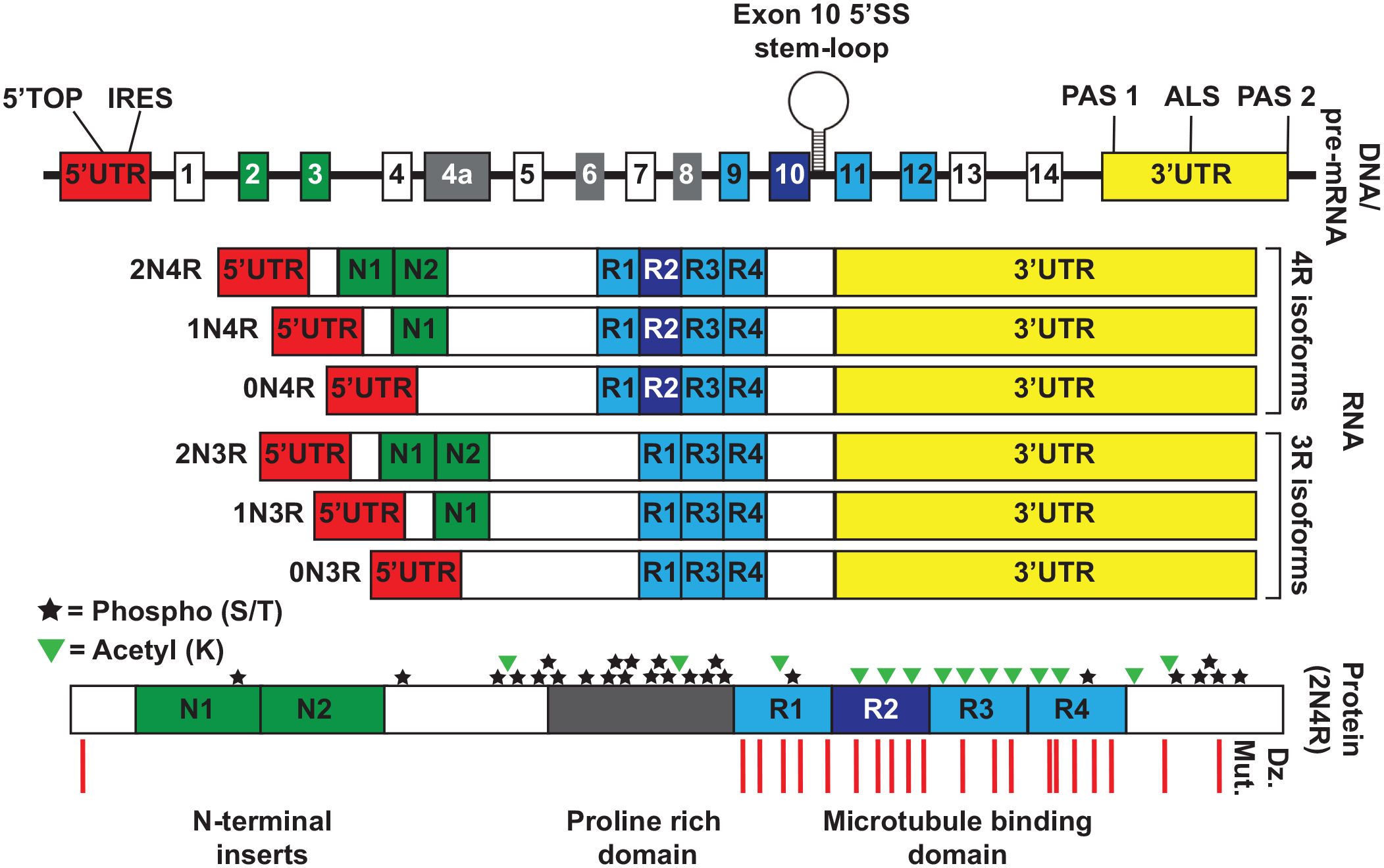
Tau structure, regulatory elements, posttranslational modifications, and disease-causing mutations. The top DNA/pre-mRNA track shows a schematic of the microtubule-associated protein tau genomic loci. The exon 10 5′SS stem-loop designates the location of several disease-causing mutations that alter the RNA splicing of exon 10. PAS 1 and PAS 2 represent polyadenylation sites that can alter the length of the 3′UTR attached to the tau mRNA. ALS in the 3′UTR refers to the location of a predicted axonal localization signal that has been observed in rat tau but not yet confirmed in the human mRNA. The RNA track shows the six splice isoforms of tau that are expressed in the human central nervous system, which differ in their inclusion or exclusion of exons 2, 3, and 10. These isoforms can have either the short or long 3′UTR. The protein track shows the location of phosphorylation and acetylation posttranslational modifications as well as disease-causing mutations (Dz. Mut.) on the full-length 2N4R tau isoform.
The 3R and 4R tau isoforms produce different types of tau fibrils, and propagation experiments have suggested that there is a seeding barrier between 3R and 4R tau fibrils where isoform pairing between seed and monomer is required for efficient propagation (Woerman and others 2016). Certain diseases have deposition of 3R tau (e.g., Pick disease), 4R tau (e.g., corticobasal degeneration [CBD] and progressive supranuclear palsy), or their combination (e.g., AD and chronic traumatic encephalopathy; Goedert 2018). Consistent with this, splicing changes that favor the 3R isoforms result in the deposition of primarily 3R tau fibrils (Anfossi and others 2011), and splicing changes that favor 4R isoforms result in the deposition of primarily 4R tau fibrils (Spillantini and others 1997; Yasuda and others 2000).
Neurodegenerative diseases that are due to mutations in tau are categorized as primary tauopathies (Kovacs 2017). Two groups of mutations in tau have been shown to drive neurodegeneration. The first group of mutations are missense mutations (primarily in the MTBD) that stabilize aberrant folding of tau and thereby promote fibrillization (e.g., P301L, P301S, G303V; Goedert and Jakes 2005; Stanford and others 2003; Figure 2).
The second group of primary tauopathy–causing mutations in tau alters the inclusion or exclusion of exon 10 and controls the ratio of 3R:4R tau. RNA splicing of tau exon 10 changes with age. Generally, there is more 3R during development, and 4R expression increases with age until a roughly 1:1 balance of 3R:4R tau is reached during adulthood (Kosik and others 1989).
Since 3R and 4R isoforms form distinct fibrillar structures (Falcon and others 2018; W. Zhang and others 2020) and can inhibit the fibrillization of the other isoform, an imbalance in the 3R or 4R form is anticipated to increase the propensity of tau aggregation (Boyko and others 2020).
Secondary tauopathies occur when the deposition of aggregated and hyperphosphorylated tau is downstream of another perturbation. Some of the secondary tauopathies and their upstream insults are AD (β-amyloid), chronic traumatic encephalopathy (repeated head trauma; Katsumoto and others 2019), and subacute sclerosing pan encephalitis (measles infection; Bancher and others 1996). The striking diversity of causes in the secondary tauopathies suggests that if they share a common pathologic mechanism, it is likely one that has a variety of inputs. One candidate is the overactivation of the neuroinflammatory system (Ising and others 2019).
Secondary tauopathies, which are not inherited in a mendelian manner, can be strongly influenced by the “neuronal microenvironment” (Figure 3). Like the concept of a “cancer microenvironment,” the neuronal microenvironment refers to the chemical composition of the extracellular environment created by neurons and glial cells that influences disease progression. Various stressors, including trauma, infection, and amyloid-β, can promote glial cells to secrete inflammatory molecules that likely influence the formation of tau aggregates in neurons, possibly through activation of the NLRP3 inflammasome (Ising and others 2019). While the exact mechanisms are still being uncovered, it is possible that inflammatory molecules can promote tau aggregation through the formation of RNA and protein assemblies named cytoplasmic speckles (CSs), which serve as preferred sites of tau aggregation. A critical aspect of tauopathies is understanding the specifics of the neuronal microenvironment and how it promotes the formation of tau aggregates.
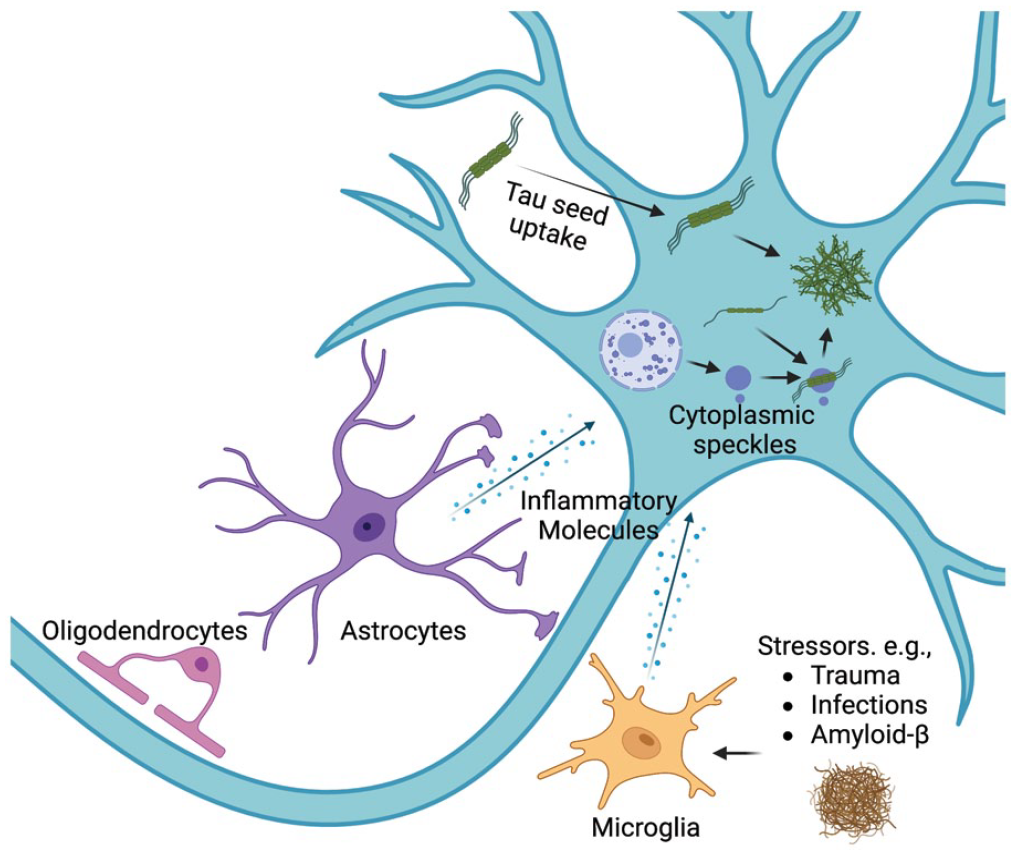
The neuronal microenvironment and tau aggregation. Tauopathies can have diverse causes, including trauma, infections, and amyloid-β. One model for how these stressors induce tauopathies is by inducing glial cells to secrete inflammatory molecules and create an inflammatory “neuronal microenvironment.” These inflammatory molecules likely trigger several changes in neurons that promote tau aggregation, including inducing the formation of cytoplasmic speckles that can serve as preferred sites of tau aggregation in neurons.
Tau Interacts with RNAs In Vitro and in Cells
Several observations demonstrate that tau binds RNA. This was first suggested by early observations showing that tau binds RNA in vitro (Bryan and others 1975). Subsequent studies have shown that tau can bind to >30-nt RNA homopolymers, tRNAs, yeast total RNA, and total human RNA with in vitro gel shift assays and biolayer interferometry (Wang and others 2006; Zhang and others 2017; Zwierzchowski-Zarate and others 2022). Truncated forms of tau containing only the proline-rich domain or the MTBD were able to bind RNA, suggesting that tau has two regions capable of binding RNA (Wang and others 2006). Since experiments in vitro show that tau can bind a range of RNAs from tRNAs to homopolymers (Lodish and others 2000; Zhang and others 2017; Zwierzchowski-Zarate and others 2022), it remains to be determined if tau has specific RNAs that it binds and regulates in neurons or other cell types.
In cells, unaggregated tau has been shown to interact with RNAs through PAR-iCLIP (photoactivatable ribonucleoside-enhanced individual-nucleotide resolution ultraviolet cross-linking immunoprecipitation). These iCLIP experiments from HEK293 cells and in human induced pluripotent stem cell (iPSC)–derived neurons suggested that tau has a preference for small structured RNAs, including tRNAs and, to a lesser extent, small nucleolar RNAs (snoRNAs; Zhang and others 2017).
Consistent with tau-binding RNA, several observations suggest that tau aggregates in model systems and disease contexts contain RNA. First, tau aggregates in AD, Down syndrome, Pick disease, progressive supranuclear palsy, and CBD stained positive for acridine orange, a dye that changes its fluorescent properties when bound to RNA and confirmed by using RNase to erase the signal (Ginsberg and others 1997; Ginsberg and others 1998). Similarly, phosphorylated tau aggregates in the muscle of patients with inclusion body myositis contain RNA (by acridine orange) and the RBP survival of motor neuron (Broccolini and others 2000). Second, RNA sequencing of purified tau aggregates from HEK293 tau aggregation models or mice with P301S-driven tauopathy has shown that tau aggregates contain a diverse transcriptome enriched for small nuclear RNAs (snRNAs) and snoRNAs and modestly enriched for Alu RNAs and tRNAs (Lester and others 2021). Consistent with this set of sequencing results, previous immunohistochemistry and immunogold labeling of tau fibrils in postmortem AD brain samples also stained positive with an antibody to the 2,2,7 trimethylguanosine cap, which is present on snRNAs and snoRNAs (Hales and others 2014; Jia and others 2007).
Could RNA Be an Important Cofactor for Tau Aggregation in Disease?
Two types of observations suggest that the interaction of tau with RNA might promote the formation and propagation of tau fibers in disease. First, RNA is one of several polyanions that can promote the initiation and propagation of tau fibrillization in vitro (Dinkel and others 2015; Friedhoff and others 1998; Kampers and others 1996). When transfected into cells, RNA-induced tau fibrils are seeding competent and capable of inducing tau aggregates in recipient cells (Zwierzchowski-Zarate and others 2022). Moreover, RNA-induced tau fibrils display toxicity in HEK293 and iPSC neuronal cell culture models, similar to tau seeds isolated from patients with tauopathies (Ash and others 2021; Oakley and others 2021; Sanders and others 2014). Finally, transfection of exogenous tRNA into cells with tau seeds leads to increased tau aggregation (Zhang and others 2017).
An unanswered question is how different RNAs might alter tau fiber formation. Seeding competent tau fibrils form most efficiently with total human RNA, and 40nt poly(A) RNA appears to constitute a minimal fibril-inducing sequence (Zwierzchowski-Zarate and others 2022). However, since fibrilization can be induced in vitro with other polyanions, including heparin, some sulfated glycosaminoglycans, or unsaturated free fatty acids (Goedert and others 1996; Lövestam and others 2022; Wilson and Binder 1997), it remains to be established if RNA is a cofactor for tau fiber growth in neurons. Given the diversity of tau fiber forms, one possibility is that different cofactors, including various RNA species, could induce different folds in tau and thereby enhance the formation of multiple tau fiber types.
A second key observation is that treatment with RNase A and RNase T1 (but not DNase) of soluble tau seeds from AD postmortem samples led to a ~70%–85% reduction in their seeding capacity in a HEK293 tau biosensor model (Zwierzchowski-Zarate and others 2022). Together these experiments suggest that RNA could play a critical role in maintaining tau seeding competence in soluble tau aggregates isolated from the brains of patients with AD.
Insight into how RNA might interact with tau fibers comes from a recent cryo-EM structure of RNA-induced tau fibrils created in vitro. This analysis revealed that RNA is bound to the positively charged residues Arg406 and His407 and aligns parallel to the fiber axis (Abskharon and others 2022). These fibrils created with total RNA isolated from mouse liver also disassemble with RNase A treatment. Notably, this in vitro structure does not precisely match fibers isolated from the brains of patients with AD; however, it is possible that this structure could represent an early step in the formation of tau fibrils (Abskharon and others 2022; Fitzpatrick and others 2017). Similarly, a cryo-EM study showed that tau fibrils formed in vitro with poly(A) RNA differed from AD and chronic traumatic encephalopathy filaments (Lövestam and others 2022). Since different RNA sequences can alter the seeding properties of the resulting fibrils (Zwierzchowski-Zarate and others 2022), one anticipates that the cryo-EM structures of tau fibrils will also differ depending on the origin and sequence of the RNA used. An important caveat to these cryo-EM studies is that the structures are often created from a fraction, often <15% of particles on the grid, and could represent one of many fibril structures present (Abskharon and others 2022; Lövestam and others 2022). Important questions going forward are whether RNA can be identified in fibril structures isolated from disease contexts and whether tau fibrils and oligomers from other tauopathies in addition to AD are sensitive to RNase.
Normal and Aberrant Interactions of Tau with RBPs
In addition to interacting with RNA, tau has been observed to interact with RBPs in normal and pathologic conditions. For example, soluble tau has been seen to copurify with RBPs (Gunawardana and others 2015). In addition, several studies have examined the protein composition of tau aggregates in model systems and in the brains of tauopathy and age-matched control patients by using a variety of techniques: 1% sarkosyl fractionation followed by liquid chromatography–tandem mass spectrometry, immunoprecipitation–mass spectrometry, laser capture microdissection–mass spectrometry, and engineered ascorbate peroxidase–mass spectrometry (Table 1).
RNA-Binding Proteins Identified through Proteomics Studies.
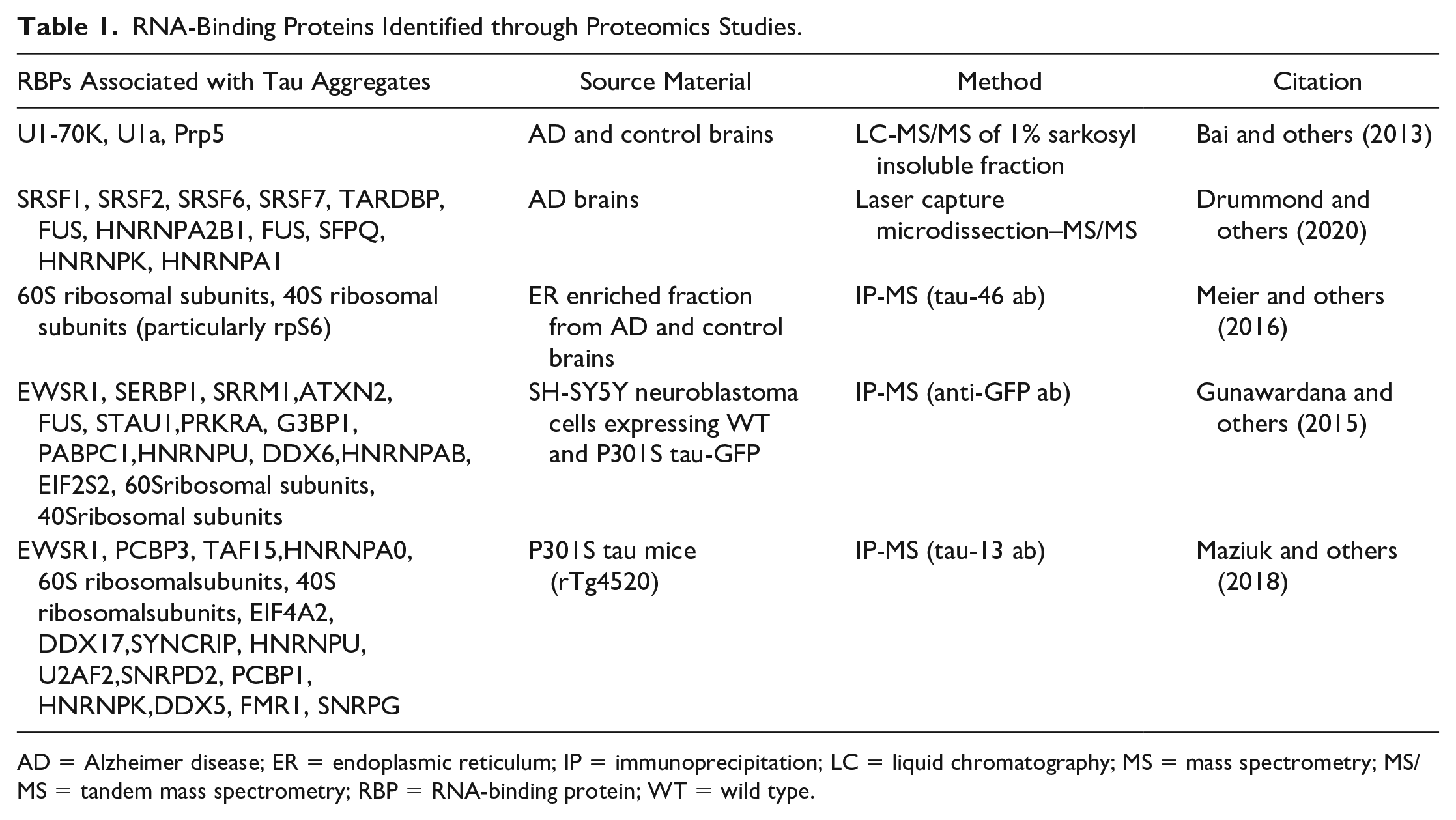
AD = Alzheimer disease; ER = endoplasmic reticulum; IP = immunoprecipitation; LC = liquid chromatography; MS = mass spectrometry; MS/MS = tandem mass spectrometry; RBP = RNA-binding protein; WT = wild type.
Some consistent themes have emerged from these investigations. First, RBPs involved in RNA splicing are often enriched in tau aggregates. For example, two proteins in the U1 snRNP (U1-70K and U1A) and Prp5, a splicing RNA helicase, are specifically enriched in the insoluble fraction (1% sarkosyl) from AD brains as compared with age-matched controls or other tauopathies (Bai and others 2013). Importantly, the enrichment of U1-70K in cytoplasmic aggregates has been confirmed in AD postmortem samples by immunohistochemistry and immunogold transmission electron microscopy (Bai and others 2013; Bishof and others 2018; Diner and others 2014; Hales and others 2014; Hales and others 2016). The enrichment of snRNP proteins is notable given the corresponding enrichment of snRNAs in tau aggregates in model systems and postmortem tissues (Hales and others 2014; Lester and others 2021).
Other RBPs involved in RNA processing that have been observed in tau aggregates include SRRM2 (a core component of the spliceosome that localizes to nuclear speckles), PNN (a binding partner of SRRM2; Lester and others 2021; McMillan and others 2021; Tanaka and others 2018), LSM4 (involved in processing and degradation of mRNA; Tracy and others 2022), HNRNPA2B1 (involved in splicing of pre-mRNA and acts as a reader of N6-methyladenosine–modified microRNAs in the nucleus; Alarcón and others 2015; Jiang and others 2021), Musashi 1/2 (nuclear localized and regulates translation of target mRNAs; Montalbano and others 2019; Montalbano and others 2020), SFPQ (involved in pre-mRNA splicing; Drummond and others 2020; Kavanagh and others 2022; Ke, Ke, and others 2012; Younas and others 2020), and numerous serine/arginine-rich splicing factor proteins (Table 1; Drummond and others 2020). A recent meta-analysis of 12 proteomics studies also showed the consistent incorporation of RBPs in tau aggregates, particularly HNRNPs (Kavanagh and others 2022).
In a study of 29 AD samples and 7 age-matched controls, SRRM2 was depleted from the nucleus and relocalized to the cytoplasm in the hippocampi and amygdala in all the AD samples but none of the age-matched controls (McMillan and others 2021). Similar nuclear depletion of SRRM2 was seen in six patient brains: three with CBD and three with FTD (Lester and others 2021). Interestingly, loss-of-function mutations in SRRM2 result in a neurodevelopmental disorder supporting its role as a protein that is critical to neurologic physiology (Cuinat and others 2022). Strikingly, the accumulation of SRRM2 and PNN in tau aggregates in model systems is determined by specific polyserine domains, which might be related to the formation of tau fibers under some conditions (Lester and others 2023). Loss of U1-70K, SRRM2, and SFPQ function (and likely other unidentified proteins) in the nucleus could contribute to the toxicity of tau aggregates by inappropriately altering RNA processing.
Several studies suggest that tau can interact with 40S and 60S subunits (Gunawardana and others 2015; Koren and others 2020; Maziuk and others 2018; Meier and others 2016; Tracy and others 2022). Immunofluorescence in AD brains has validated these studies and showed colocalization between rpS6 (40S subunit) and tau oligomers (Meier and others 2016). Similarly, tau was observed interacting with ribosomes >30 years ago by electron microscopy (Nelson and others 1993; Papasozomenos and Binder 1987). In one immunoprecipitation–mass spectrometry study, the authors treated their samples with and without RNase and observed a loss of many tau-ribosome interactions, suggesting that these are mediated by RNA (Gunawardana and others 2015). It is hypothesized that tau’s interaction with rpS6 leads to impaired translation of 5′TOP RNAs that code for ribosomal proteins (and likely other stress response proteins), exacerbating tau pathology (Koren and others 2019).
The interaction of soluble or aggregated tau with RBPs and the ribosomes raises two important implications. First, this suggests that tau might have a normal function in the regulation of RNA physiology in yet-to-be-discovered manners. Second, these observations suggest that the sequestration of RBPs in tau aggregates might alter the normal function of these proteins and lead to alterations in RNA processing, thereby contributing to the toxicity of tau aggregates. Whether tau is involved in regulating RBPs under normal physiologic conditions is unknown but is an intriguing possibility.
How Could RNA or RBPs Affect Tau Aggregation?
An important and yet-to-be-resolved issue is understanding the mechanism by which the interactions of tau with RNA and RBPs might affect tau aggregation. In principle, RNAs or RBPs could interact with tau and stabilize a folded state of the monomer that is more prone to fiber formation (Figure 4A). Conversely, such interactions could stabilize a folded monomer state that is inhibited for fiber formation. Alternatively, larger RNAs could bind multiple tau molecules and create a higher local concentration that might promote the initiation of tau fibrillization (Figure 4B). However, no direct data for these mechanisms have been presented.
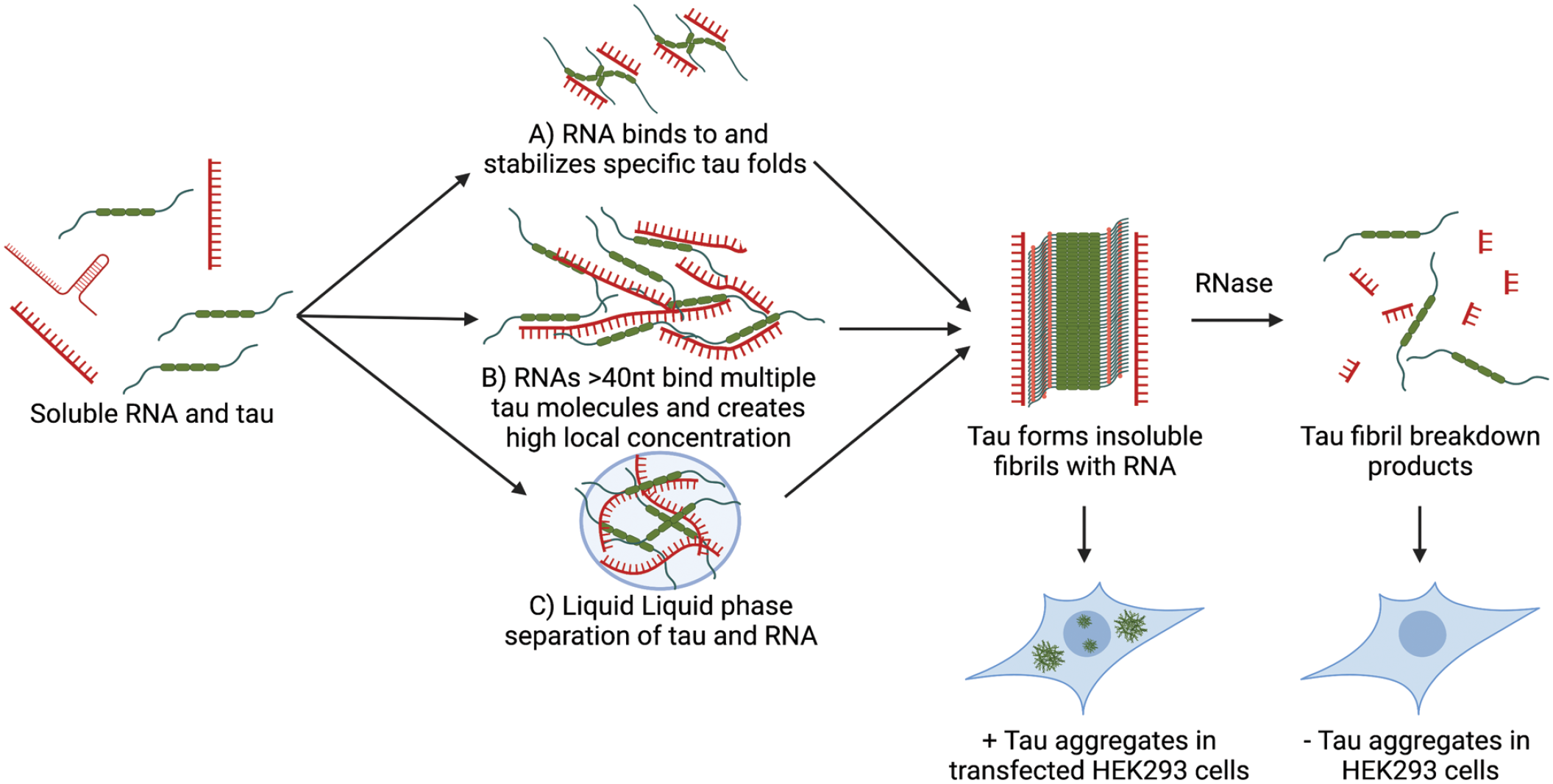
In vitro interactions between tau and RNA. Tau binds RNA in vitro, and this interaction can promote the formation of tau fibrils. We envision three nonmutually exclusive mechanisms by which RNA could promote the formation of tau fibrils. (A) Tau binds to tau monomers or dimers and stabilizes a specific fold that can form larger tau fibrils. (B) RNAs >40 nt could bind to multiple tau monomers at either the microtubule-binding domain or the proline-rich domain, thereby creating a high local concentration of tau and promoting stochastic fibrilization. (C) Liquid–liquid phase separation of RNA and tau promotes a high local concentration of tau leading to fibrilization. Once tau fibrils have formed, cryo–electron microscopy structures have suggested that RNA is bound parallel to the fibril axis as shown. RNase treatment of tau fibrils reduces seeding capacity when transfected into biosensor cell lines and a loss of fibril integrity as seen by transmission electron microscopy.
An emerging possibility is that tau interacting with RNA or specific RBPs might promote a larger assembly that concentrates tau through a process referred to as liquid–liquid phase separation (LLPS) and thereby promotes tau aggregation (Figure 4C; Ambadipudi and others 2017; Ash and others 2021; Boyko and others 2020; Ferreon and others 2018; Kanaan and others 2020; Ukmar-Godec and others 2019; Ukmar-Godec and others 2020; X. Zhang and others 2020). LLPS is a phenomenon defined as the metastable demixing of proteins and/or RNAs mediated by multivalent interactions (Hyman and others 2014; Van Treeck and Parker 2018). LLPS has been shown to concentrate proteins and promote the fibrillization of several proteins involved in neurodegeneration, including FUS and hnRNPA1 (proteins involved in RNA processing; Lin and others 2015; Moleix and others 2015; Patel and others 2015).
Tau undergoes LLPS in vitro when mixed with polyanions such as RNA (Hochmair and others 2022; Lin and others 2019) or heparin (Lin and others 2020) or when mixed with crowding agents such as polyethylene glycol (Ambadipudi and others 2017). Similarly, tau has been suggested to undergo LLPS in conjunction with the TIA1 RBP (Ash and others 2021). The high local concentration of tau in the dense phase of an LLPS might increase rates of fiber initiation and propagation. In agreement with this idea, mixtures of tau:tRNA resulted in LLPS assemblies that were salt-sensitive, SDS-resistant, and seeding-competent tau aggregates. When a molecular crowding agent, polyethylene glycol, was added to these tau:tRNA mixtures, a gel-like substance was produced at physiologic salt concentrations (Hochmair and others 2022).
Other reports suggest that LLPS is not a prerequisite for the fibrilization of tau and that the two are actually separate processes that merely occur under similar in vitro experimental conditions (Lin and others 2020). Thus, whether LLPS of tau and RNA or some other membraneless organelle creates a high local concentration of tau to promote tau fibrillization in cells remains to be established.
Do RNA and Protein Granules Affect Tau Aggregation?
Several membraneless organelles composed of RNA and protein, collectively referred to as RNP granules, have been observed to interact with tau. These include nuclear speckles, mitotic interchromatin granules (MIGs), and the related CSs, as well as some components of stress granules (SGs; Figure 5). In agreement with tau’s enrichment in splicing related RNAs and RBPs, nuclear tau aggregates were observed to form in nuclear speckles in cell and mouse models of tauopathies (Lester and others 2021). Nuclear speckles are a membraneless organelle thought to play roles in transcription, splicing, and export of nascent mRNAs and contain a high concentration of snRNAs, RNA processing and export factors, and transcription machinery (Lester and others 2021; Spector and Lamond 2011). Tau aggregates disrupt the composition, organization, and dynamics of nuclear speckles, which may be related to altered patterns of pre-mRNA splicing in cells harboring tau aggregates (Lester and others 2021). This disruption in pre-mRNA splicing by tau aggregates may be related to the splicing alterations seen in analyses of postmortem brain tissue from patients with tauopathies (Apicco and others 2019; Hsieh and others 2019; Raj and others 2018).
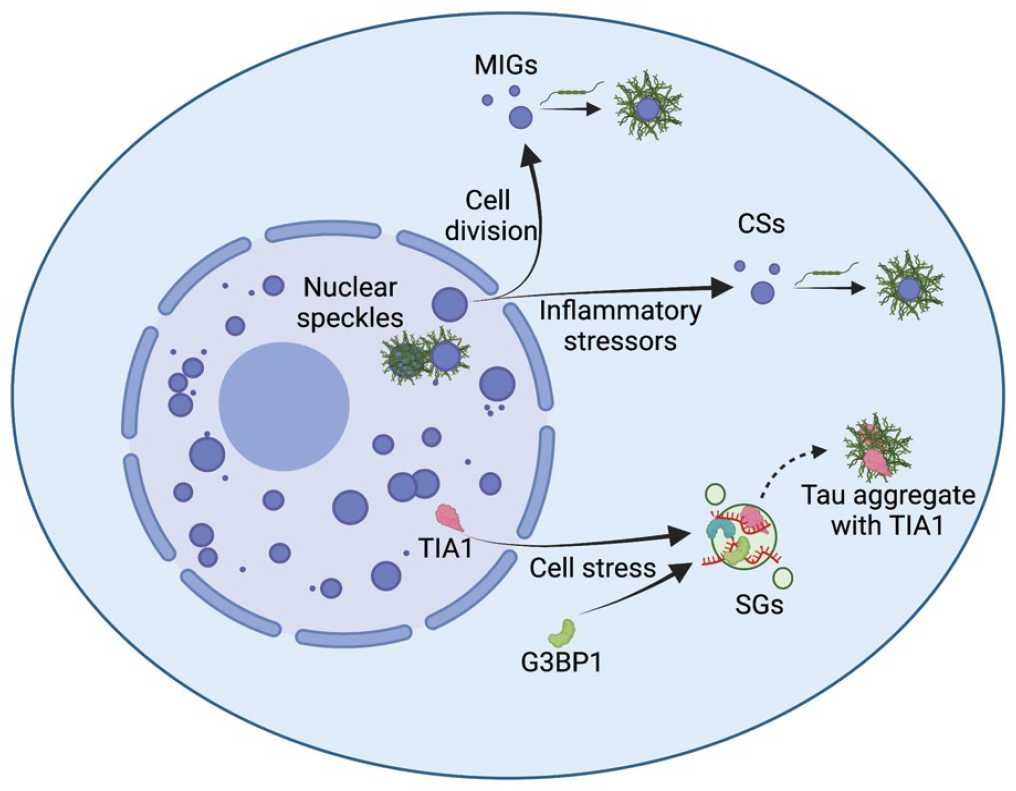
Interactions between tau and membraneless organelles. Tau has been observed to interact with several membraneless organelles that could promote the conversion of monomeric tau to aggregates of tau, including mitotic interchromatin granules (MIGs), cytoplasmic speckles (CSs), and stress granules (SGs). MIGs form during cell division when the nuclear membrane breaks down. CSs have been observed to form in response to inflammatory stressors and inhibition of nuclear import. SGs form in response to various cellular stressors, and while canonical SGs form distinct structures from tau aggregates, some components of SGs have been observed to colocalize with tau aggregates, namely TIA1.
Several components of nuclear speckles, including SRRM2 (a core component of the spliceosome), relocalize to cytoplasmic tau aggregates in model cell line and mouse systems (Figure 5; Lester and others 2021). Importantly, this mislocalization of proteins occurs in human disease since SRRM2 has been observed to mislocalize to cytoplasmic tau aggregates in three human tauopathies (AD, CBD, and FTD tau; Lester and others 2021; McMillan and others 2021). In cell line models, a polyserine domain is necessary and sufficient for the mislocalization of the nuclear speckle proteins SRRM2 and PNN to tau aggregates (Lester and others 2023). A parsimonious model is that the polyserine domains also target SRRM2 to tau aggregates in human tauopathies.
Recent evidence argues that the formation of cytoplasmic RNP granules containing SRRM2 and presumably other nuclear speckle components can serve as preferred sites of tau fiber growth and thereby increase tau aggregation. Time-lapse imaging of HEK293 tau biosensor cells expressing a tagged version of the endogenous SRRM2 protein revealed that tau aggregates will associate with and grow from the surface of cytoplasmic SRRM2-containing assemblies (Lester and others 2023). SRRM2 is known to form cytoplasmic assemblies during mitosis, referred to as MIGs (Xu and others 2022), and can form similar assemblies independent of mitosis in cell lines and neurons, referred to as CSs (Berchtold and others 2018; Lester and others 2023; Tanaka and others 2018).
Interestingly, the movement of SRRM2 from the nucleus to the cytoplasm and the phosphorylation of SRRM2 occur early in AD and in the 5XFAD mouse model of AD (Tanaka and others 2018). This has been suggested to occur due to endoplasmic reticulum stress activating Erk1/2, which then phosphorylates SRRM2 at Ser1068 and leads to SRRM2 accumulation in the cytosol (Tanaka and others 2018). The accumulation of SRRM2 in the cytoplasm leads to the formation of SRRM2 assemblies that can subsequently serve as sites for tau aggregation (Lester and others 2023). The preferred growth of tau fibers in association with MIGs or CSs argues that there is a specific biochemical feature of these assemblies that enhances tau fiber growth in cells. Future work should focus on the mechanisms by which amyloid-β and other inflammatory stressors trigger endoplasmic reticulum stress and Erk1/2 activation, leading to SRRM2 phosphorylation and relocalization to the cytoplasm.
An emerging possibility is that proteins containing polyserine domains are a critical factor sufficient to enhance tau fiber growth. This is suggested by the observation that overexpression of polyserine domains leads to the nucleation of polyserine assemblies that are sufficient to serve as preferred sites of tau fiber growth in HEK293 biosensor cells (Lester and others 2023). Moreover, overexpression of polyserine domains in HEK293 cells increases tau aggregation, while knockdown of the polyserine domain in the highly expressed PNN protein reduces tau aggregation (Lester and others 2023). Whether polyserine acts directly to promote tau fiber growth or there are required modifications and/or cofactors that contribute to this growth will require additional experiments.
Two observations suggest that the formation of CSs in neurons could be relevant to human disease. First, the accumulation of toxic amyloid-β aggregates can trigger specific phosphorylation of SRRM2, leading to its accumulation in cytoplasmic assemblies (Tanaka and others 2018). Second, in human iPSC-derived cortical neurons, endogenous inflammatory compounds such as PJD2 and PGE2 induce the formation of CSs (Lester and others 2023). Important areas for future work will be investigating the mechanisms governing CS formation in postmitotic neurons and whether altering the formation of such CSs alters tau aggregation.
SGs are another membraneless organelle that have been associated with the formation of tau aggregates (Figure 5). SGs are mRNA-protein assemblies formed from nontranslating mRNAs and associated proteins that form in response to cellular stress that inhibits translation (Protter and Parker 2016). SG components, such as TIA1 and eiF3n, have been shown to colocalize with tau aggregates in a tauopathy mouse model (Apicco and others 2018; Vanderweyde and others 2016). Tau aggregates form distinct structures from canonical G3BP1 containing SGs, and the RNA composition of tau aggregates does not resemble that of SGs, suggesting that tau may be interacting with a subset of canonical SGs (Khong and others 2017; Lester and others 2021; Lester and others 2023; Vanderweyde and others 2012). Additionally, heterozygous knockout of TIA1 is protective against tauopathy in mice, suggesting a role for TIA1 in the toxicity of tau aggregation (Apicco and others 2018).
Interestingly, RNA and protein assemblies have been shown to change and condense with age in the brains of Drosophila (Pushpalatha and others 2022). As aging is the primary risk factor for neurodegenerative tauopathies such as AD and FTD, it is possible that RNPs lose their fluidity with time, leading to greater stability and abundance. Whether CSs, MIGs, or SGs also change with age and how such changes affect tau aggregation is unknown.
Genetic Interactions of RBPs and Tau Toxicity
Two RBPs have been shown to reduce tau toxicity in mouse models when mutated. In one example, a tauopathy mouse model heterozygous for TIA1 and expressing the pathogenic human P301S tau showed reduced toxicity and extended life span (Apicco and others 2018). The reduced level of TIA1 is proposed to increase the formation of NFTs and reduce the formation of more neurotoxic oligomers (as assessed by reaction with “oligomer-specific” TOC1 antibody), possibly due to direct interactions between TIA1 and tau (Ash and others 2021).
A second example comes from a screen in Caenorhabditis elegans that identified loss-of-function mutations in SUT-2 (suppressor of tauopathy 2) that rescued the toxicity of human tau expression (Guthrie and others 2009). Importantly, PS19 tau mice lacking the mammalian homolog (MSUT2, ZC3H14) also show reduced phosphorylated tau pathology, suggesting that this protein acts similarly in mammalian systems (Wheeler and others 2019). MSUT2 localizes to nuclear splicing speckles where it binds poly(A) RNA and plays a role in the control of poly(A) tail length in neuronal cells (Guthrie and others 2011; Kelly and others 2014). The CCCH finger domains of MSUT2 mediate its interaction with RNA and are responsible for promoting tauopathy (Wheeler and others 2019). In contrast to MSUT2, knockdown of PABPN1—a protein that acts in opposition to MSUT2 and increases mRNA poly(A) tail length—exacerbates tau pathology (Wheeler and others 2019). This suggests that shorter poly(A) tails may enhance tau pathology via an unknown mechanism. Mutations in MSUT2 can lead to intellectual disability (Pak and others 2011), and levels of MSUT2 are decreased in patients with AD, suggesting a possible role for MSUT2 in human pathophysiology (Wheeler and others 2019).
Toxicity of Tau Aggregation and Possible Connections to RNA Physiology
A poorly understood and critical aspect of tauopathies is understanding the basis for neurotoxicity due to the assembly of tau into aberrant forms. One unresolved issue is whether toxicity is due to smaller oligomeric tau assemblies, which in some assays can be more toxic than larger fibers (Ghag and others 2018; Mirbaha and others 2015; Zwierzchowski-Zarate and others 2022), due to consequences of the formation and buildup of larger NFTs or some combination of these two cellular perturbations, which is the most likely situation. Another key issue is whether a perturbed aspect of neuronal physiology is critical to neuronal death. Several possibilities have been suggested for tau toxicity, such as dysregulation of neuronal transport, activation of caspases, abnormal Ca2+ control, impaired mitochondrial function, and increased DNA or RNA damage (Kopeikina and others 2012; Violet and others 2014; Violet and others 2015; Zhang and others 2021). A key point is that since loss of MSUT2 (Wheeler and others 2019) or reduced levels of TIA1 (Apicco and others 2018) can suppress tau toxicity in mouse models, this raises the strong possibility that at least some aspect of tau toxicity is due to perturbations in RNA physiology.
Tau aggregation could alter RNA physiology through multiple different mechanisms (Figure 6). First, the accumulation of RNA splicing factors in tau aggregates might sufficiently deplete these factors from the nucleus to alter splicing patterns (Figure 6A). This possibility is supported by the observation that in a HEK293 tau biosensor model, inducing tau aggregation alone is sufficient to alter RNA splicing and lead to widespread intron retention (Lester and others 2021). This could be relevant since patients with AD display alterations in RNA splicing in postmortem tissues that can be observed at the level of RNA expression and at the level of protein expression (Apicco and others 2019; Hsieh and others 2019; Johnson and others 2018; Raj and others 2018). Alternatively, the accumulation of tau aggregates in nuclear speckles with corresponding changes to their organization and dynamics might also alter splicing (Figure 6B; Lester and others 2021). Future studies that make use of short- and long-read RNA sequencing of tau knockout models should provide more information to the role of tau in RNA metabolism in normal and pathologic conditions.
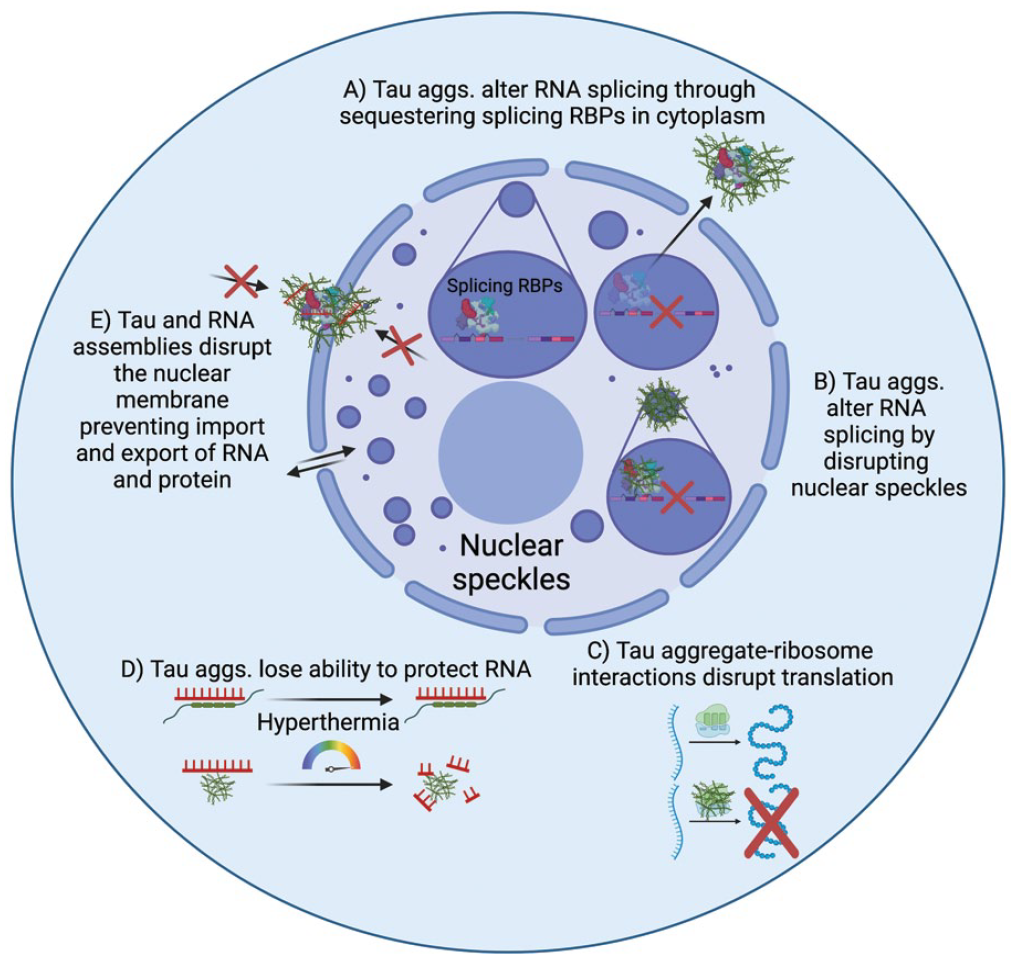
Possible mechanisms of tau aggregate toxicity related to RNA and RNA-binding proteins (RBPs). In healthy cells, RNA splicing takes place in nuclear speckles where splicing-related RBPs, including U1-70K and SRRM2, are located. (A) Tau aggregates have been observed to colocalize with splicing-related RBPs in the cytoplasm, and depletion of these splicing factors from the nucleus is expected to result in disrupted RNA splicing. (B) Similarly, tau aggregates have been observed to form within nuclear splicing speckles and disrupt their organization, dynamics, and composition, possibly also contributing to RNA splicing defects. (C) Tau aggregates can bind ribosomal subunits and alter the translation of mRNAs through global inhibition of translation or by altering which mRNAs are translated. (D) Tau has been proposed to stabilize RNA and DNA. In tau knockout mice and tauopathy mice, there is greater RNA degradation in response to hyperthermic conditions. (E) Tau aggregates can disrupt the nuclear membrane and the ability of RNAs and proteins to transit through nuclear pores.
Tau has been shown to affect protein translation, and this effect is thought to be mediated by binding to ribosomal subunits (Figure 6C; Koren and others 2019). Tau fibers have been shown to hinder translation in vitro, and tau associates strongly with ribosomes in vivo (Table 1). Interestingly, tau seems to bias which mRNAs are translated through binding to and reducing the activity of rpS6, a ribosomal subunit that promotes the translation of 5′TOP mRNAs coding for ribosomal and translational machinery (Koren and others 2019).
Loss of function of tau due to its assembly in aggregates might alter RNA stability (Figure 6D). Tau knockout mice have been used to study the phenotypic changes in response to tau loss of function. One proposed physiologic role for tau’s interaction with RNA is that tau protects RNA from oxidative damage under physiologic and hyperthermic conditions (Violet and others 2014). In agreement with this hypothesis, tau KO mice subjected to heat shock (44 °C for 20 min) showed increased TUNEL staining of RNA and DNA fragments relative to wild type mice (Violet and others 2014). Similarly, mice expressing the G272V and P301S tau mutations showed an increase in TUNEL staining (terminal deoxynucleotidyl transferase dUTP nick end labeling) of RNA and DNA fragments with heat shock (Violet and others 2015). Tau aggregation possibly abrogates this function, leading to increased DNA and RNA damage.
While possibly contributory, loss of function is unlikely to be the primary mechanism of tau toxicity, as there are few reported disease-causing loss-of-function mutations in tau, and tau knockout models show little to no neurodegenerative phenotype (Ke, Suchowerska, and others 2012).
Another emerging possibility is that tau and RNA assemblies disrupt the nuclear pore complex, preventing cells from importing proteins or exporting mRNAs that are essential for normal functioning (Figure 6E; Eftekharzadeh and others 2018; Prissette and others 2022). In agreement, a CRISPR-Cas9 screen for modifiers of tau aggregation found that knockout of genes involved in the maintenance and repair of the nuclear envelope led to greater tau aggregation in a HEK293 tau biosensor model (Prissette and others 2022). Interestingly tau aggregation leads to the formation of nuclear envelope invaginations that accumulate polyadenylated RNAs (Cornelison and others 2019). Together these studies suggest that tau aggregation can disrupt the nuclear membrane and that disruptions in the nuclear membrane can lead to greater tau aggregation. How these mechanisms interact and how RNA becomes trapped in these nuclear defects remains to be determined.
Concluding Remarks
Insoluble fibrillar aggregates of tau are seen in many neurodegenerative diseases. Despite significant research interest, there are still numerous questions about how tau aggregates form, the effect that tau aggregates have on cellular physiology, and tau’s normal physiologic function. Interactions between RNA and RBPs with tau, as well as RNA’s ability to promote the formation of tau fibrils in vitro, imply a role for RNA in the development and toxicity of tau aggregates (Figure 6). RNA and protein assemblies, particularly MIGs and CSs, have emerged as a possible mechanism by which tau aggregates encounter RNA and RBPs (Figure 5). This raises the possibility that manipulating the formation and stability of RNA and protein assemblies could alter tau aggregate formation. Critical issues to address in future work are whether tau has any normal role in regulating RNA physiology and whether the accumulation of RNA and RBPs in tau aggregates promotes their formation or toxicity in patients.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: R.P. is a founder and consultant for Faze Medicines.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institutes of Health (NIH; 1F30AG063468-04 to E.L., 5R01GM045443-31 to R.P.) and the Howard Hughes Medical Institute (to R.P.)