
Abstract
Fatally injured neurons may necrose and rupture immediately, or they may initiate a programmed cell death pathway and then wait for microglial phagocytosis. Biochemical and histopathologic assays of neuronal death assess the numbers of neurons awaiting phagocytosis at a particular time point after injury. This number varies with the fraction of neurons that have necrosed vs initiated programmed cell death, the time elapsed since injury, the rate of phagocytosis, and the assay’s ability to detect neurons at different stages of programmed cell death. Many of these variables can be altered by putatively neurotoxic and neuroprotective interventions independent of the effects on neuronal death. This complicates analyses of neurotoxicity and neuroprotection and has likely contributed to difficulties with clinical translation of neuroprotective strategies after brain injury. Time-resolved assays of neuronal health, such as ongoing expression of transgenic fluorescent proteins, are a useful means of avoiding these problems.
Preventing neuronal death is a fundamental goal linking diverse areas of translational neuroscience. While many studies have reported neuroprotective interventions that reduce neuronal death in acute and chronic neurologic disorders, these interventions have translated very poorly to clinical practice (O’Collins and others 2006; Schmidt-Pogoda and others 2020). The causes of this translational failure are not known, but a great deal of thought has gone into possible etiologies.
As with any unexpected experimental result, the first step in understanding the failure of translation of basic neuroprotection studies is to examine the scientific methods employed. This approach was also suggested by the poor reproducibility of many translational findings in other fields (Ioannidis 2005; Prinz and others 2011). Consequently, increases in the rigor of scientific inquires have been widely adopted as a means to increase replicability and, ideally, translation to clinical practice. Improvements in blinding, randomization, number of replications, heterogeneity of subjects, and statistical analyses are some of the important interventions that have been proposed to improve reproducibility (Fisher and others 2009; Landis and others 2012; Lapchak and others 2013; Stroke Therapy Academic Industry Roundtable 1999). However, a corresponding increase in clinical translation has not yet been observed (Chamorro and others 2021; National Academies of Sciences, Engineering, and Medicine 2019; Schmidt-Pogoda and others 2020). This begs the question: what other factors may be contributing to the failure to translate experimental neuroprotective strategies to human care?
Here we consider a separate barrier to the successful translation of basic neuroprotection research: the experimental quantification of neuronal death. Assays of neuronal death are central to all neuroprotection research, yet the process of cell death is complex and only partly understood. Assays of cell death are generally based on one element of the death process (Fricker and others 2018; Galluzzi and others 2009). The element of the death process that drives the assay may be altered by a putatively neuroprotective intervention, but this does not guarantee that there is an accompanying change in the rate of cell death. If there were no accompanying change in the death rate, the effects of the intervention on the assay would create false-positive or false-negative effects of the putative neuroprotective intervention, depending on the direction of the effect of the intervention on the assay. A related problem is that the biological processes that drive the assays are themselves complex. The sensitivity and specificity of the assay can change with experimental conditions (Bouchier-Hayes and others 2008; Macklis and Madison 1990; Zille and others 2012). This again may result in spurious effects on the rate of neuronal death, with consequent false-positive or false-negative effects of the putative neuroprotective intervention. We begin with some background information regarding cell death pathways and then illustrate errors that can arise from the complexity of programmed neuronal death. We then discuss errors arising from the interaction of experimental conditions and the assays used to measure neuronal death.
It Ain’t Over ’til It’s Over: Programmed Neuronal Death
Necrosis—also known as oncosis and accidental neuronal death—refers to cell death occurring as a consequence of catastrophic energy failure (Fricker and others 2018; Vanden Berghe and others 2010). It is widespread in the core of cerebral infarcts and in the setting of severe global hypoxic ischemic injury (Zille and others 2012). Characteristics include ATP depletion, calcium influx, cell swelling, and cytoplasmic membrane rupture with rapid dispersion of nuclear and cytoplasmic contents (Vanden Berghe and others 2010). Necrosis initiates rapidly at the time of energy failure (Barros and others 2001; Northington and others 2001; Williams and others 1991). Prevention of necrosis has not been a focus of neuroprotective interventions, primarily because the rapid temporal evolution of necrosis renders neuroprotective intervention largely infeasible for injury-related, or primary, necrosis (Galluzzi and others 2009).
In contrast, apoptosis—a form of regulated, or programmed, cell death—refers to an active form of neuronal death in which the cytoplasmic membrane does not rupture so that cellular contents are not released to the extracellular space (Bertheloot and others 2021; Galluzzi and others 2009). Apoptosis begins with activation of proteases that dismantle the protein synthetic capacity of the cell, and it ends in the phagocytosis of the dying neuron by microglia. The ordered death of apoptosis proceeds along a time scale that is much longer than necrosis (Damisah and others 2020; Ferriero 2002; Lee and others 2019; Nakajima and others 2000; Perego and others 2011). The decision to enter the apoptotic pathway is based on either intrinsic or extrinsic triggering events. Early events in apoptosis include the activation of caspases (Logue and Martin 2008; Namura and others 1998) and other proteases that destroy the nucleic acids and protein synthetic machinery of the neuron, including ribosomal complexes (DeGracia and Hu 2007). The fate of the neuron is thus sealed at this early time point because there is no way for neurons to recover from the loss of ability to synthesize proteins (Thilmann and others 1986; Widmann and others 1991). Apoptosis proceeds through the removal of the neuron’s dendritic inputs (Johansen and others 1984; Neigh and others 2004) to the final end point of phagocytosis (Fricker and others 2018; Mike and Ferriero 2021; Stolzing and Grune 2004). There are many other important pathways of programmed cellular death that we have not touched on. This minireview focuses on apoptosis and necrosis because these are the principal pathways of neuronal death after acute brain injury (Fricker and others 2018; Moskowitz and others 2010).
If necrosis and apoptosis result in the same end point, death of the neuron, what is the point of the more complex process of apoptosis? Apoptosis maintains the physical integrity of the cytoplasmic membrane, although in the absence of protein synthesis the membrane becomes more leaky, or permeable, over time (Puig and others 2018). The intact membrane keeps the cytoplasmic contents, including damage-associated molecular patterns and high concentrations of the excitatory neurotransmitter glutamate, out of the extracellular space, where these contents could lead to damage or destruction of neighboring neurons (Bertheloot and others 2021). An analogy for the role of neuronal apoptosis vs necrosis is staged academic retirement vs quitting suddenly (Figure 1). The function of the university is maintained more smoothly if a professor engages in a staged retirement that provides the time for the recruitment of a successor, graduation of mentored students, and distribution of resources such as laboratory space. Yet, if the professor quits suddenly, these processes must be executed more haphazardly, with a substantial possibility of disadvantaging the professor’s students and colleagues.
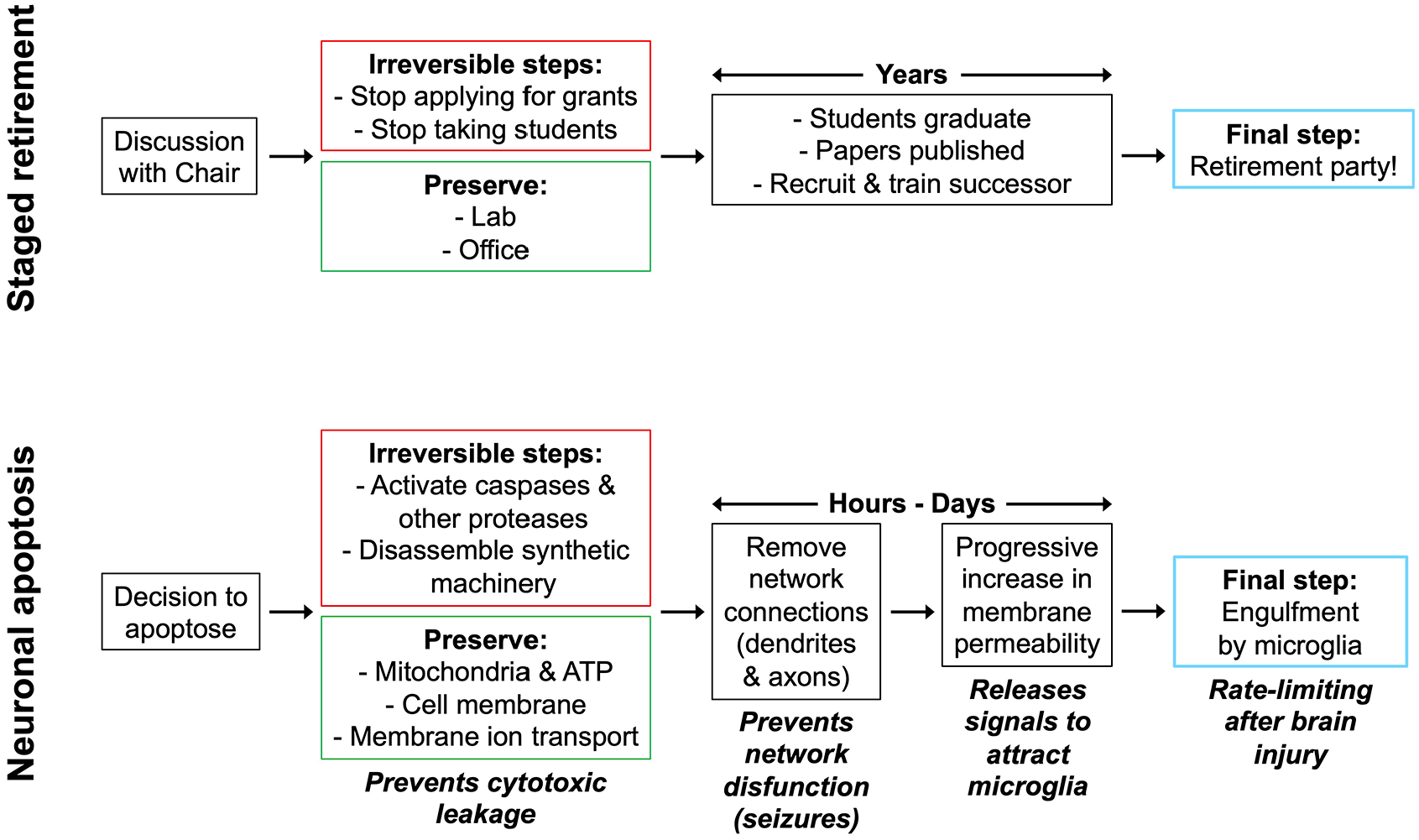
Programmed cell death as the “stage retirement” of neurons. The upper panel summarizes the staged retirement of a research professor. The corresponding lower panel summarizes neuronal apoptosis, or programmed neuronal death.
Similarly, if a neuron dies after being disconnected from the network, the network function is much less affected than if the neuron undergoes terminal depolarization while its synaptic connections are still intact and capable of releasing transmitter in response to the terminal depolarization (Andrew and others 2022; Hartings and others 2003; Puig and others 2018). Spilling of the cytoplasmic contents due to oncotic membrane rupture would harm neighboring neurons and glial cells (Choi and Rothman 1990), whereas keeping cytoplasmic contents within the cytoplasm until they can be phagocytosed by microglia is less damaging and anti-inflammatory (Chen and Trapp 2016; Poon and others 2014).
Timing Is Everything
Although microglia make haste to find dying neurons (Elliott and Ravichandran 2016; Kamei and Okabe 2023), there is no guarantee that an activated microglial cell will be available at the precise moment when a neuron initiates apoptosis. This is particularly true when large numbers of neurons die at the same time, as may occur after brain injury (Adamchik and others 2000; Berdichevsky and others 2012; Bonde and others 2002; Conti and others 1998; Du and others 1996; Dzhala and others 2012; Ferriero 2002; Lee and others 2019; Li, Chopp, Jiang, Yao, and Zaloga 1995; Li, Chopp, Jiang, and Zaloga 1995; Morrison and others 2006; Northington and others 2001; Pozzo Miller and others 1994; Strasser and Fischer 1995). Under these conditions, a neuron may need to remain in the queue for phagocytosis until a microglial cell can migrate to the neuron and phagocytose it (Adamchik and others 2000; Bonde and others 2002; Damisah and others 2020; Ferriero 2002; Lee and others 2019; Morrison and others 2006; Nakajima and others 2000; Perego and others 2011; Pulsinelli and others 1982; Strasser and Fischer 1995; Figure 2). This delay has been exploited for over a century by neuropathologists and neuroscientists, who have devised many histocytologic methods to count the number of neurons awaiting phagocytosis (Bouchier-Hayes and others 2008; Galluzzi and others 2009; Jones and Senft 1985; Schmued 2016; Segawa and Nagata 2015; Switzer 2000; van Engeland and others 1998; Zhang and others 2018).
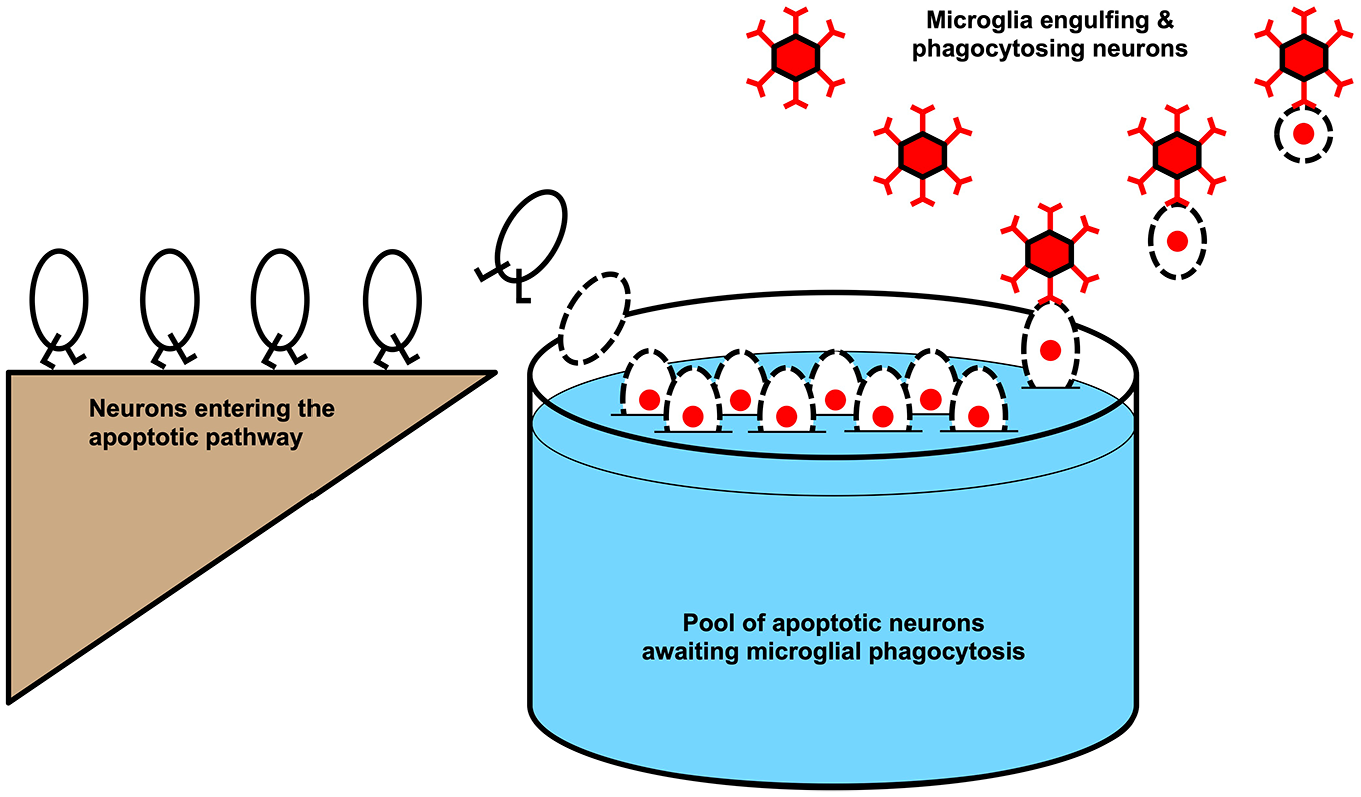
The pool of apoptotic neurons awaiting microglial phagocytosis. Neurons are ovals; dotted lines represent permeable membranes; red nuclei denote propidium iodide (PI) staining; and the red hexagons are microglia that remove neurons from the pool. The neurons in the pool are drawn as PI positive so that this is the same pool of neurons that would be visualized by a PI stain. This pool of neurons grows steadily larger when more neurons are entering it or fewer neurons are being removed by microglia.
Why would neuropathologists and neuroscientists be interested in the number of neurons awaiting phagocytosis? This number has long been used to estimate the rate of neuronal death (Berdichevsky and others 2012; Ikonomidou and others 2000; Pozzo Miller and others 1994; Sullivan and others 2002). From this imputed rate of death, various additional inferences are then made regarding the severity of the injury that incited the death, as well as the efficacy of any neurotoxic or neuroprotective interventions designed to alter the rate of death.
But a fundamental question is this: how well does the number of neurons awaiting phagocytosis (Figure 2) reflect the rate of neuronal death after an insult? In choosing a checkout line in the grocery store, we never assume that the longest line got that way because customers are preferentially queuing there. Rather, we assume that the checker is slow; we also assume that the shortest line is that way because it is being served by the fastest checker. However, when we look at the queue of neurons awaiting apoptosis, we make the opposite assumptions: that the neurons are being phagocytosed, or checked out, at equal rates and that the length of the queue therefore directly reflects the rate at which neurons are entering it. If we then make a neurotoxic or neuroprotective intervention, we look at the length of the queue of neurons awaiting phagocytosis (i.e., the number of neurons in the pool in Figure 2) and make the further assumption that the change in the length of the queue reflects the impact of the intervention on the rate of neuronal death, not the rate of phagocytosis.
How valid is the assumption that the rate of neuronal death can be measured by the length of the queue of apoptotic neurons awaiting phagocytosis? The grocery checkout line analogy suggests that the rate of phagocytosis should be considered as well as the rate of neuronal death. It is the difference in the rate of neuronal apoptosis initiation (whereby neurons enter the queue) and the rate of phagocytosis (whereby neurons exit the queue) that alters the length of the queue.
As it turns out, the rate of neuronal phagocytosis is not at all constant. The rate of neuronal phagocytosis is variable and can be altered by many conditions that are canonically considered neurotoxic or neuroprotective. For example, it is universally accepted that epileptic seizures are neurotoxic, particularly when they are prolonged (Meldrum and Brierley 1973; Nevander and others 1985). However, it is quite difficult to prove this point conclusively with human or experimental data (Bozarth and others 2019; Hesdorffer and others 1998; Nishiyama and others 2015; Raspall-Chaure and others 2006; Stafstrom and Holmes 2002). Seizures are triggered by abnormal conditions such as brain injury or the experimental application of convulsants, and these proconvulsant conditions themselves may be neurotoxic (Bozarth and others 2019; Meldrum and others 1973; Nevander and others 1985; Srivastava and others 2020; Vespa and others 2007). Could it be that experiments demonstrating the neurotoxicity of seizures were measuring alterations in the rate of phagocytosis of neurons killed by the initial ictogenic insult, rather than alterations in the effect of seizures themselves on the rate of neuronal death?
To phagocytose a neuron, microglia must be activated and then may need to migrate to the neuron that is to be phagocytosed. To find the neuron, microglia sense exocrine signals released by the dying neuron. These signals are conveniently termed “find me” and “eat me” signals and are composed of substances such as chemokines and purines that activate their cognate receptors on microglia (Boada-Romero and others 2020; Butler and others 2021; Pontejo and Murphy 2021; Yu and others 2022). However, these molecules are also released during neuronal activity and in great amounts during seizures (Beamer and others 2021; Cerri and others 2016). The release of these compounds by multiple viable neurons makes it much harder for microglia to find apoptosing neurons. Consequently, neuronal activity in general slows down microglial migration (Grinberg and others 2011). Could seizure activity also slow down microglial phagocytosis?
We evaluated the role of seizures in neuronal apoptosis using organotypic hippocampal slice cultures. This preparation is prepared at postnatal day 7 by culturing acute hippocampal slices. Slicing the brain causes a lot of neuronal trauma and death (Bak and others 1980; Dzhala and others 2012); in addition, there is ongoing neuronal apoptosis in the hippocampus at this age (Eyo and others 2016). Thus, microglia in the organotypic slice preparation already have their hands full phagocytosing dying neurons. In the normal-developing brain, developmental apoptosis is also occurring at P7, but it is less marked than in the recently traumatized (sliced) organotypic hippocampal cultures (Eyo and others 2016; Lossi and others 2009). This makes the organotypic slice preparation a very useful preparation for studying programmed neuronal death after injury.
When we assessed the effect of seizures on neuronal death, we initially assayed single time points using propidium iodide (PI) to identify neurons in the apoptotic queue. We found that seizures increased the size of the queue and blocking seizures reduced the size of the queue (Berdichevsky and others 2012). We initially concluded that seizures were neurotoxic by their effect on the size of the apoptotic queue. However, seizures also slow down the rate of microglial phagocytosis dramatically (Abiega and others 2016; Balena and others 2023). Conversely, blocking seizures with anticonvulsants accelerates phagocytosis (Abiega and others 2016; Balena and others 2023; Figure 3).
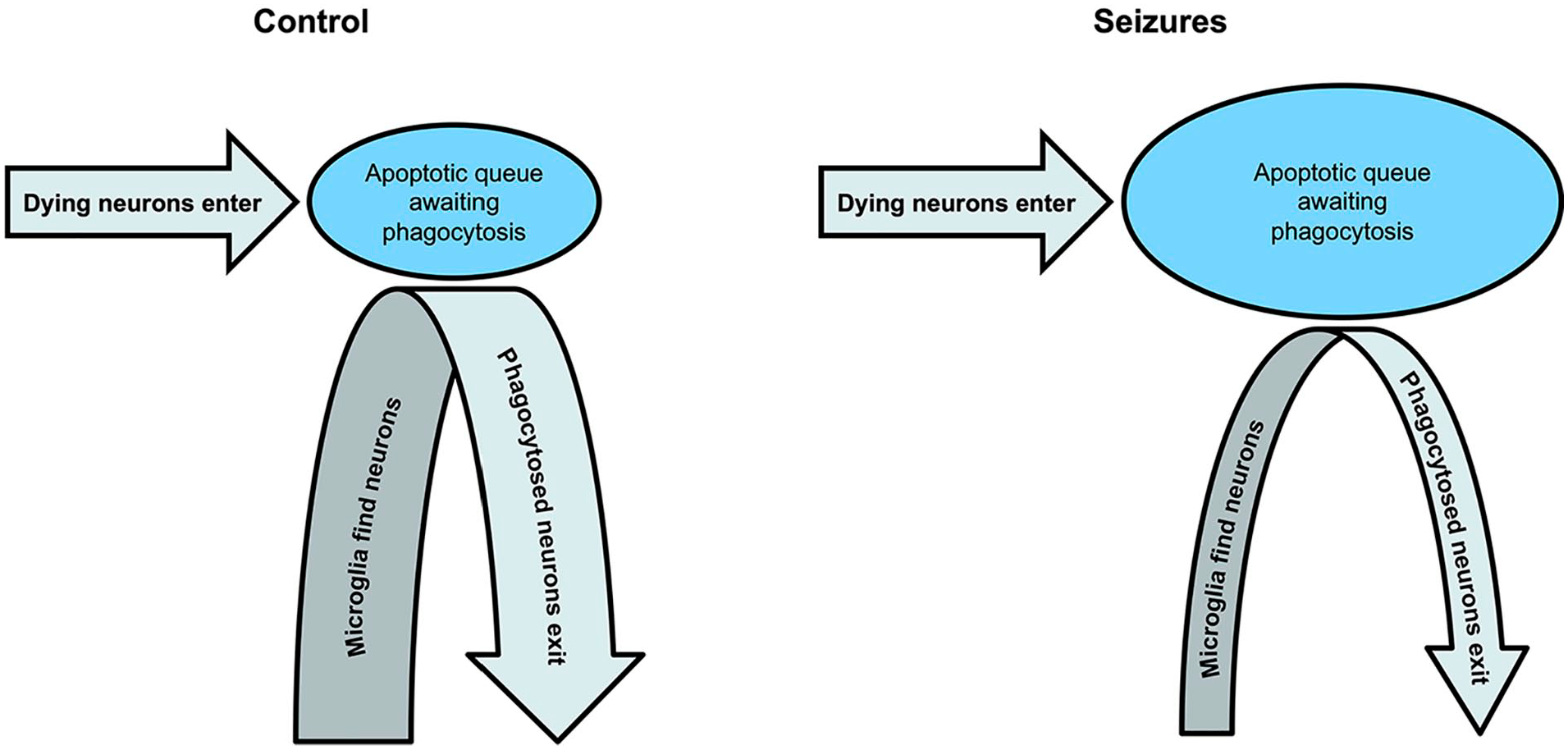
Seizures slow microglial migration to dying neurons, resulting in a backlog of neurons awaiting phagocytosis. The enlarged queue gives the appearance of a neurotoxic effect because more dying neurons are visible with vital dyes. Yet, the number of apoptotic neurons entering the queue over time has not changed.
The slowing of microglial phagocytosis by seizure activity greatly complicates the logic underlying the conclusion that seizures are neurotoxic. If the rate of phagocytosis is reduced by seizures, then assays of neurotoxicity based on the length of the queue of neurons awaiting phagocytosis will reflect not only changes in the rate of cell death but also changes in the rate of phagocytosis. How can these two variables be separated?
To address this problem, we turned to time-resolved assays of neuronal death (Figure 4). We tested whether seizures, or arrest of seizures with glutamate antagonists, injured neurons that were healthy enough to express a transgenic fluorescent protein. We again used the in vitro organotypic hippocampal brain slice preparation, which makes feasible the transgenic labeling of neurons, as well as serial observations of the number of neurons in the queue awaiting phagocytosis. It has been established that once neurons commit to programmed cell death, the emission from these fluorescent proteins is rapidly quenched (Arrasate and Finkbeiner 2005; Arrasate and others 2004; Balena and others 2023; Linsley and others 2019; Steff and others 2001; Strebel and others 2001). The mechanism of fluorescent protein quenching has not been established but is considered to be a consequence of 1) enhanced bleaching of the fluorescent protein by cytoplasmic reactive oxygen species (Alvarez and others 2010; McLean and others 2009) released during the mitochondrial permeability transition (Zorov and others 2014) and 2) destruction of protein synthetic capacity in programmed cell death (Clemens and others 2000; Thilmann and others 1986), which prevents the synthesis of unbleached fluorescent proteins.
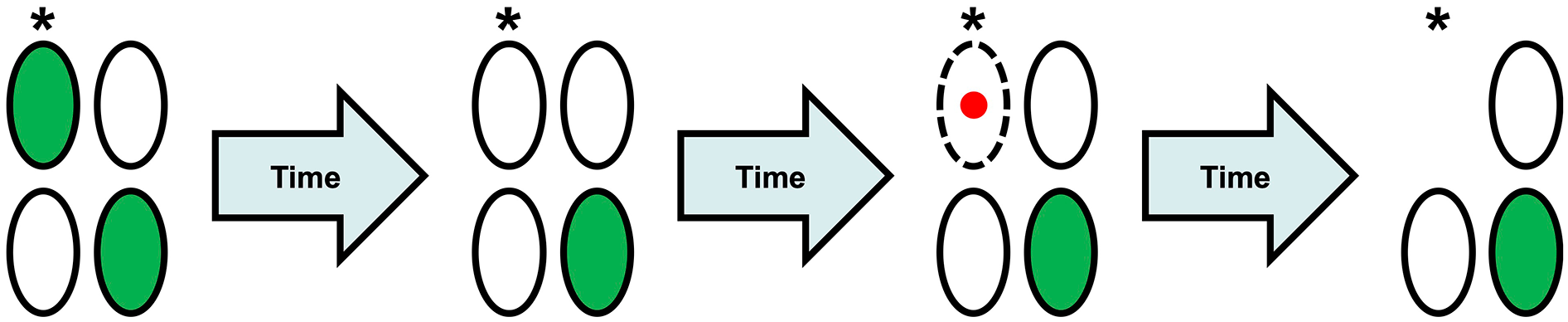
Fluorescent protein (FP) quenching during programmed cell death. Neurons are ovals; dotted lines represent permeable membranes; and red nuclei denote propidium iodide (PI) staining. Serial observations of the same population of neurons allow for the visualization of apoptosis. Upon initial observation, some neurons (but not all) are FP positive, such as the neuron indicated with an asterisk (*). As hours or days pass, a neuron that was initially FP positive will often quench. After even more time passes, that neuron’s membranes will become sufficiently permeable to allow the admittance of cell death stains, such as PI. Finally, the neuron will be engulfed and phagocytosed by microglia.
Surprisingly, repeated seizures had no effect on the health of neurons expressing transgenic proteins. However, blocking seizures with a broad-spectrum glutamate antagonist resulted in significant neuronal injury, assayed by the quenching of fluorescence of transgenic fluorescent proteins in individual neurons. Note that the glutamate antagonist blocked all glutamatergic transmission, not just seizures, and blocking all synaptic activity is likely to induce apoptosis in developing neural networks (Heck and others 2008; Ruijter and others 1991; Schonfeld-Dado and others 2009; Wong and others 2018). Similarly, if glutamate receptors are activated with sufficient intensity and duration, neurotoxic effects can be induced (Abele and others 1990). Nevertheless, it is remarkable that repeated seizures had no demonstrable neurotoxic effect when assayed by transgenic fluorescent protein emission. This suggests that the apparently neurotoxic effect of seizures in the same preparation, when assayed by the number of neurons in the queue awaiting phagocytosis (Berdichevsky and others 2012), was due to alterations in the rate of microglial phagocytosis (Abiega and others 2016). In the grocery store checkout line analogy, the seizures were slowing down the checkers and thereby extending the checkout line, rather than altering the rate at which shoppers joined the line (i.e., the rate at which neurons committed to apoptosis and joined the queue awaiting phagocytosis).
The alteration of phagocytosis rates by purportedly neurotoxic measures (seizures) or neuroprotective ones (i.e., anticonvulsants) is one example of how a seemingly straightforward result can be wrong, such as an increase in PI-positive dying neurons in a seizing preparation. Seizures are not the only purportedly neurotoxic condition that might result in spurious measures of neuroprotection and neurotoxicity. Most acute injuries to the brain, including hypoxic-ischemic injury and trauma, result in acute necrosis of the most injured neurons—with the release of many chemotactic signals, or damage-associated molecular patterns, that microglia use to reach the neurons in penumbral areas (i.e., less severely compromised) that are undergoing programmed cell death (Gülke and others 2018; Shao and others 2022). This flood of signals from ruptured necrotic neurons is likely to confuse microglia in the same manner that seizures do, resulting in similar reductions in phagocytosis of neurons undergoing programmed neuronal death.
Of course, wrong results will not translate to clinical practice. Arguably, this somewhat arcane effect is not likely to represent a significant cause of failure of translation; nevertheless, the entire subfield of neuroprotection via reductions in excitotoxicity has failed to translate (O’Collins and others 2006; Schmidt-Pogoda and others 2020), and it is certainly possible that effects such as those previously described contribute to this failure.
Another problem related to the change in the rate of phagocytosis of dying neurons is the timing of the assay for dying neurons relative to injury. If the difference in the rate of entry into the process of apoptosis vs the rate of exit by phagocytosis alters the size of the queue of dying neurons, then the amount of time that the rates have differed is also critical. Imagine a boat that has sprung a leak, and the rate of water influx is larger than the rate at which the water can be bailed out. As such, whether the boat is afloat or sunk is a function not only of the difference in rates but also the time that has elapsed since the leaking and bailing began. In the grocery store analogy, the longer the slow checker works, the longer that line will become relative to the faster checkers, as long as the rate of shoppers joining the queues is equal. Similarly, the queue of dying neurons will steadily expand if the rate of neuronal entry into the apoptosis pathway exceeds the phagocytosis rate. Likewise, the queue will steadily shrink if the rate of phagocytosis exceeds the rate of entry into the apoptotic process. The actual size of the queue will depend on the difference in the rates and the amount of time that the rates have differed. As an example, assaying stroke volume three days after stroke produces much more heterogenous results than assays conducted six hours after stroke (Unal Cevik and Dalkara 2003; Zille and others 2012). One important contributor to this variance will be this difference in the rate of neuronal death vs phagocytosis, the impact of which steadily increases with time.
To make things more complex, the rate of neuronal entry into apoptosis and the rate of microglial phagocytosis can change over time as well as with experimental manipulations. A brief surge in neuronal apoptosis due to injury may not be reflected in the number of neurons in the queue for phagocytosis if too much time is allowed between injury and assay of the queue length. In that time interval, sustained phagocytosis can remove the excess neurons. We found evidence that the rate of phagocytosis increases with the number of neurons in the queue awaiting phagocytosis (Balena and others 2023). A pulse of PI labels a cohort of apoptotic neurons in the queue awaiting apoptosis. With serial imaging, we found that the number of labeled neurons declined monoexponentially with time, indicating that the rate of phagocytosis varied with the number of neurons in the queue. However, continued entry of newly apoptotic neurons into the queue may have reduced the rate of microglial phagocytosis of PI-labeled neurons, so this issue requires additional investigation.
A final complication is that neurons that are in the queue for apoptosis, particularly if subject to another insult, may undergo necrosis rather than wait for apoptosis. This process is often referred to as secondary necrosis (Vanden Berghe and others 2010). Similar to the departure of impatient shoppers from the longest queue, the necrotic neurons disappear from the apoptotic queue, causing the queue to shorten. In assays based on the size of the apoptotic queue, such as PI labeling, this reduction in the number of apoptotic neurons would give a false-positive neuroprotective signal. All these effects make the size of the queue of neurons awaiting phagocytosis very difficult to interpret solely in terms of the rate of entry into the apoptotic queue (i.e., the rate of neuronal death), but that is the current practice. Time-resolved, or sequential, assays of neuronal death avoid these problems and provide much more accurate measures of neuronal death (Arrasate and others 2004; Balena and others 2023; Linsley and others 2019).
The Invisible Neuron
Besides PI, how is the size of the queue of apoptotic neurons awaiting phagocytosis measured? Since the 19th century, neurons undergoing apoptosis but not yet phagocytosed have been visualized by biomarkers that can be quantified by light microscopy (Galluzzi and others 2009; Zille and others 2012). For example, when stained with a combination of the two textile dyes hematoxylin and eosin, neurons in the process of apoptosis appear shrunken and preferentially stain with eosin; the basis for this preferential staining is not known. Vital dyes such as trypan blue, silver salts, and PI are polar dyes that preferentially stain neurons in the process of apoptosis (Bouchier-Hayes and others 2008; De Olmos and Ingram 1971; Jones and Senft 1985; Macklis and Madison 1990; Schmued 2016; Switzer 2000; Unal Cevik and Dalkara 2003). Counting the number of stained neurons in a defined volume of brain tissue provides an estimate of the length of the queue of neurons in the process of apoptosis and awaiting phagocytosis.
Vital dyes do not stain healthy neurons because the healthy cytoplasmic membrane is less permeable to polar dyes (Bouchier-Hayes and others 2008; De Olmos and Ingram 1971; Jones and Senft 1985; Macklis and Madison 1990; Schmued 2016; Switzer 2000). This brings up a critical point: although apoptosis is distinguished from necrosis by the presence of an intact cytoplasmic membrane, the permeability of the cytoplasmic membrane of apoptotic neurons increases over time (Balena and others 2023; Unal Cevik and Dalkara 2003). This progressive deterioration of the cytoplasmic membrane is likely due to a lack of spare parts: the synthetic machinery of the neuron is destroyed at the onset of the apoptotic process. As a consequence, the membrane potential depolarizes and calcium levels rise in the apoptotic neuron, although the levels are far from the levels that drive necrosis (Balena and others 2023; Macklis and Madison 1990).
The deterioration of the cytoplasmic membrane is a necessary step for staining apoptotic neurons with vital dyes. Vital dyes are also referred to as exclusion dyes because they are excluded from and do not stain healthy cells. We found that more permeable dyes, such as those conjugated to aminomethoxyester (AM) moieties (Tsien 1981), selectively permeate apoptotic neurons from the earliest resolvable points in apoptosis—that is, at the time that fluorescent proteins quench and caspase indicators such as FLICA become positive (Balena and others 2023; Figure 5). Astrocytes, microglia, and healthy neurons expressing transgenic fluorescent proteins were not labeled. This makes sense because the AM moiety is designed to facilitate the membrane permeation of dyes such as the calcium-sensitive FURA series or the sodium sensitive indicator SBFI. However, even the AM moiety does not enable permeation across healthy neuronal membranes without additional manipulations to increase membrane permeability, such as local pressure or detergents (Hamad and others 2015; Stosiek and others 2003). In the organotypic brain slice preparation, we found that AM dyes label neurons throughout apoptosis, from the time of caspase activation and fluorescent protein extinction points through phagocytosis (Balena and others 2023).
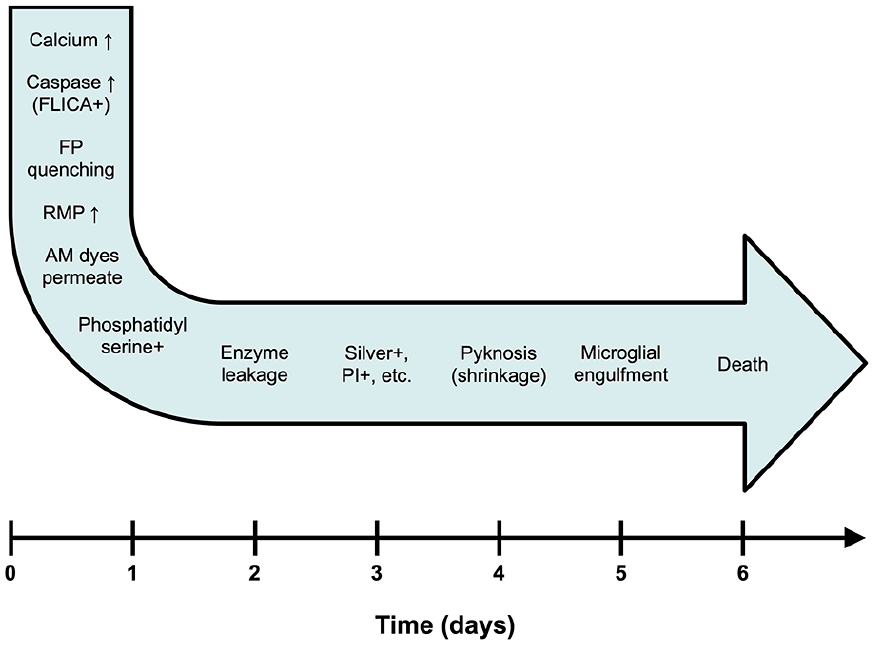
Identifying apoptotic neurons over the time course of apoptosis. Different biomarkers become positive at different stages of apoptosis. In the organotypic slice preparation, neurons that had committed to apoptosis spent two weeks in the queue awaiting phagocytosis (Balena and others 2023). Use of different biomarkers will identify different subpopulations of neurons in the queue. AM = aminomethoxyester; FP = fluorescent protein; PI = propidium iodide; RMP = resting membrane potential.
This pan-apoptotic labeling is unique to AM dyes and clearly does not occur with the established polar vital dyes such as PI (Balena and others 2023). We found that in the organotypic brain slice preparation, several days elapsed between the initiation of apoptosis and staining with PI. While this is perfectly in line with the gradual increase in permeability of the cytoplasmic membrane and depolarization of the membrane potential during apoptosis, it renders neurons early in the course of apoptosis invisible to vital dyes. Thus, many guidelines for staining with vital dyes recommend particular time intervals between injury and staining (Zille and others 2012). However, depending on the nature or severity of the insult, neurons may progress through stages of apoptosis at different rates (Zille and others 2012). As such, at any one time point, neurons may be in the process of apoptosis but are not yet stainable with vital dyes, or they may have progressed all the way through apoptosis and are now phagocytosed. In either case, although they have committed to a cell death program, they are invisible to the vital dye and will not be counted in the apoptotic queue or in estimates of neurotoxicity or neuroprotection based on the size of the queue.
Translational Excursion 1: Invisibility in Alzheimer Disease
The apoptotic queue is more than an exasperating experimental complication. It also illuminates a long-standing, unsolved puzzle in Alzheimer disease. Alzheimer disease is a neurodegenerative disease that is characterized by progressive brain atrophy (Sabuncu and others 2011) and neuronal loss (Mann and others 1988). Paradoxically, it is very difficult to find any neurons undergoing apoptosis in the brains of patients with Alzheimer disease (Stadelmann and others 1998). This has led to a search for other forms of neuronal death in Alzheimer disease (Mangalmurti and Lukens 2022). However, as long as the rate of neuronal apoptosis is lower than the rate of microglial phagocytosis, there should be virtually no neurons in the apoptotic queue despite cumulatively massive neuronal loss over many years. With no neurons in the apoptotic queue, there will be nothing to see when the brain tissue is examined with vital dyes postmortem.
Translational Excursion 2: Invisibility After Stroke
The increasing sensitivity of histochemical assays of cell death with time after commitment to apoptosis can also cause translational problems. Perhaps the most pressing of these involves the death of neurons in the penumbra of acute ischemic stroke. Neurons in the core of the stroke die of necrosis, and neurons outside the stroke area remain healthy. The penumbra refers to the intervening areas, where neurons do not have enough blood supply to function but may not yet have committed to either necrosis or apoptosis (Baron and others 1981; Manning and others 2014). Although neuroprotective strategies for stroke have consistently failed (O’Collins and others 2006; Schmidt-Pogoda and others 2020), the empirical development of methods to remove occlusions from major cerebral arteries has produced remarkable improvements in stroke outcome (Campbell and others 2016). These studies are characterized by inclusion and exclusion criteria that favor small necrotic cores and large penumbral regions identified by acute cerebral blood flow imaging (Shafie and Yu 2021). There is great uncertainty as to what happens in the penumbra. The prevailing thought is that penumbral neurons with inadequate blood flow are at a critical juncture and will undergo apoptosis if local blood flow is not rapidly increased (Hossmann 2012), perhaps as a consequence of the metabolic stress of repeated spreading depolarizations (Andrew and others 2022). Yet, delayed mechanical removal of the large vessel occlusions is remarkably effective, a phenomenon that is not consistent with a time-dependent expansion of the necrotic infarct (Hossmann 2012) and is currently described as the late window paradox (Albers 2018). It may be that under low-flow conditions, vessels in the ischemic penumbra eventually thrombose, leading to the death of penumbral neurons. Unfortunately immunohistochemical assays are not able to address this question because of their dependence on the time from initiation of apoptosis, which can create the impression of progressive neuronal death in the penumbra (Back and others 2004; Zille and others 2012). Functional assays such as clinical testing cannot distinguish nonfunctional but viable neurons from neurons in the apoptotic queue, and imaging studies are not able to detect apoptosis acutely. Time-resolved, longitudinal studies of the viability of neurons in the ischemic penumbra are needed to understand the basis of the improved outcomes after reperfusion.
Removing the Cloak of Invisibility From Dying Neurons
Assessing neuronal death with vital dyes is already complex, but it gets worse. As mentioned earlier, detergents help permeabilize cytoplasmic membranes to enhance dye entry into neurons (Felix 1982; Stadler and others 2010; Viryasova and others 2019; Vitale and others 1993). However, a variety of agents besides detergents can serve to permeabilize the neuronal membrane. These include dimethylsulfoxide (DMSO) and alcohols (Jones and Greenfield 1987; Komatsu and Okada 1995; Ly and Longo 2004; Scalia and others 2017). Both these agents are often used as vehicles to dissolve experimental reagents. As you might guess, this can cause a problem if DMSO, alcohol, or similar solvents are used to dissolve experimental agents that are purported neurotoxins or neuroprotectants. The problem arises because the vehicle enhances the membrane permeability of the vital dye so that neurons become visible at earlier stages of apoptosis than in control conditions. This is particularly problematic in the developing brain, where large numbers of neurons undergo apoptosis physiologically (Dekkers and others 2013). Thus, an agent dissolved in DMSO might appear to be neurotoxic in the developing brain (Bittigau and others 2002) because the visibly stained apoptotic queue becomes larger after exposure to this agent. But in fact the DMSO vehicle might simply be increasing the access of a vital dye such as PI to neurons just entering the apoptotic queue that would not normally stain with PI.
For example, ethanol is a known teratogen, and exposure during the first and second trimesters of pregnancy definitely causes human fetal alcohol syndrome (May and others 2013; Wozniak and others 2019). A compelling experimental model was developed with rodents at a much later stage of development—the day before birth (Ikonomidou and others 2000). This corresponds to the third trimester of human gestation (Luhmann and Fukuda 2020), which should have been outside the time window for the teratogenic effects of alcohol, but a robust neurotoxic effect of overnight in vivo alcohol exposure was demonstrated (Ikonomidou and others 2000) by using silver to stain degenerating neurons (De Olmos and Ingram 1971). De Olmos developed this silver staining method to stain individual degenerating axons while minimizing silver uptake into normal tissue, and so membrane permeabilization is minimal in this protocol (De Olmos and Ingram 1971). Although silver stains are unique in their ability to stain not only the soma but also degenerating neuronal processes shortly after injury (Beltramino and others 1993; Moratalla and others 2021), such staining is not visible in Ikonomidou and others 2000. This suggests that the stained neurons had not died recently—specifically, that the increase in neuronal staining by ethanol exposure was a consequence of the unmasking of neurons that were already in the apoptotic queue prior to the alcohol exposure but whose membranes were not yet sufficiently permeable to admit silver ions. We wondered whether the exposure to high levels of alcohol in vivo prior to silver staining was effectively permeabilizing the membrane of neurons in earlier stages of developmental apoptosis, making them more likely to stain with silver. Such neurons would not be visible with a dye such as silver without a prior manipulation to increase membrane permeability (Figure 6).
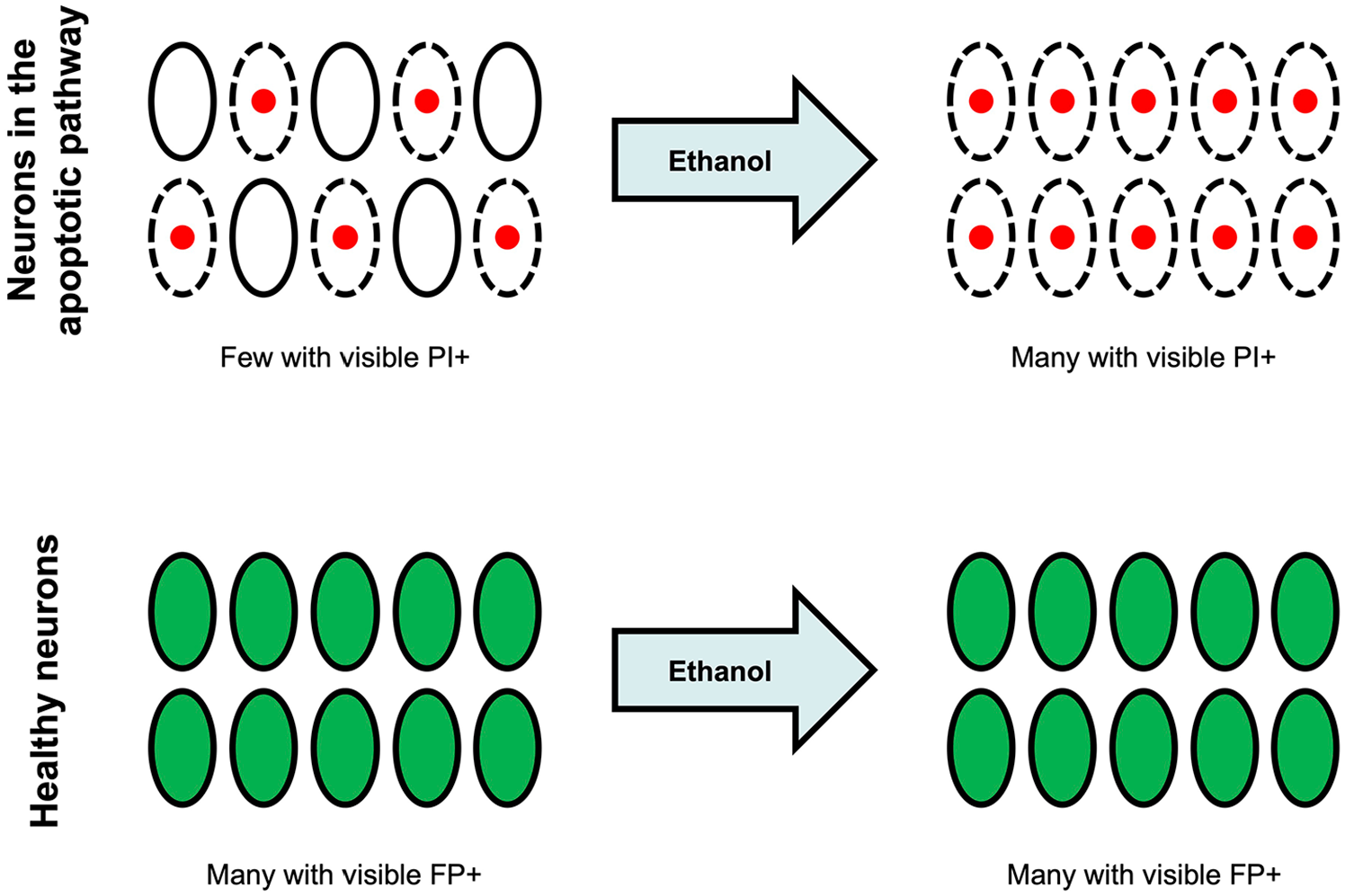
Apoptotic neurons that are too early in the process to be stained with a vital dye become stainable after treatment with a permeabilizing agent such as an alcohol. Neurons are ovals; dotted lines represent permeable membranes; red nuclei denote propidium iodide (PI) staining; and green soma denote fluorescent protein (FP) positivity. The membrane permeabilization and consequent enhanced staining with vital dyes inadvertently increase the size of the visible apoptotic queue. Healthy neurons, identified by transgenic fluorescent protein expression, are not affected by the ethanol exposure at this developmental stage. This experimental design produces a spurious neurotoxic signal for the permeabilizing agent or any drug that may have been dissolved in the permeabilizing agent.
Using the organotypic slice preparation, we found that exposure to a similar concentration of alcohol (100 mM) as the pups experienced in vivo (Ikonomidou and others 2000) resulted in enhanced staining with PI but had no effect on healthy neurons assayed by continued emission of transgenically expressed fluorescent proteins (Balena and others 2023). The most straightforward interpretation of these findings is that the alcohol is serving to increase the membrane permeability of neurons that are no longer fluorescent because they are undergoing normal developmental apoptosis. The increased membrane permeability induced by ethanol enables staining with vital dyes such as PI.
Whether a similar enhancement of membrane permeability occurs after exposure to concentrations of alcohols and DMSO used to dissolve potential neurotoxic or neuroprotective agents remains to be seen. This is an important question because many anticonvulsants and anesthetics have been tested in this model and found to be neurotoxic by virtue of an increase in the queue of apoptotic neurons awaiting phagocytosis (Aker and others 2015; Bittigau and others 2002; Noguchi and others 2017; Wise-Faberowski and others 2005). Some of these effects may be due to enhancement of membrane permeability by the vehicles used to dissolve the putative neurotoxins, with subsequent improved labeling by the vital dye. There is a large body of literature in this area, and other means of assessing apoptosis, such as TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling), and behavioral analyses have been used in these experiments (Colman and others 2023); thus, it will be important to define the role of solvents in these results. Clinical practices have been substantially altered by these experiments (Food and Drug Administration 2019), despite a lack of human data supporting cognitive injury in randomized trials (McCann and others 2019), so identifying all causes of increased staining in these exposure experiments will be an important line of inquiry.
To summarize, staining dead neurons and counting them seems like a simple and robust means of determining the neurotoxicity or neuroprotective potential of an experimental manipulation. However, the size of the visible queue of dying neurons turns out to be affected by many factors other than the number of neurons joining the queue:
Phagocytic capacity: neurons leave the queue by being phagocytosed, but the local phagocytic capacity can be overwhelmed after large-scale neuronal injuries, increasing the time that neurons spend in the queue.
Rate of phagocytosis, which varies considerably due to 1) the size of the queue and 2) the activity of neurons in the area, which may confuse the microglia and inhibit efficient microglial migration to dying neurons
Pharmacologic agents that alter the transmission or reception of “find me” or “eat me” signals sent by neurons to microglia
Conversion from apoptosis to necrosis, with rapid disappearance of the necrotic neurons
Visibility of the queue of dying neurons, which can be changed by either of the following: 1) the time in the queue, as the cytoplasmic membrane deteriorates and becomes increasingly permeable to vital dyes; 2) the application of agents as vehicles or putative neurotoxins/neuroprotectants, which alter the permeability of the membrane of apoptotic neurons and thereby the staining with vital dyes
A Better Mousetrap
In light of these problems, we have come to rely on the persistence vs quenching of fluorescence emission as a more robust measure of neuronal death (Arrasate and Finkbeiner 2005; Arrasate and others 2004; Balena and others 2023; Linsley and others 2019). Quenching of fluorescence can occur because of either necrosis or apoptosis. Thus, the contributions of both mechanisms to neuronal death are measured, although fluorescence quenching does not distinguish between these mechanisms of cell death. Fluorescence quenching requires at least two counts of fluorescent neurons: once before injury and at least once after. Quenching measures the sum of necrotic neurons and the neurons that enter the queue of apoptotic neurons awaiting phagocytosis. Because the size of the queue is not measured, the errors enumerated in the prior section will not be encountered.
Because multiple counts of neurons are required, stable microscopic access to the preparation must be available. Organotypic brain slices provide that access in vitro (Balena and others 2023). Cortical windows provide similar access in vivo (Bahari and others 2024); in vivo testing of the ideas presented here is a high priority. Fluorescence quenching also requires robust expression of a transgenic fluorophore. There is some variation in the sensitivity of different fluorophores to neuronal death, but we have generated very reproducible results with green fluorescent protein, enhanced green fluorescent protein, cyan fluorescent protein, yellow fluorescent protein, and turbo-red fluorescent protein.
We have cited our own investigation heavily (Balena and others 2023) because there are no other articles focused on this useful new approach to time-resolved analysis of neuronal demise after acute injury. However, this technique has been well characterized by other groups that have developed it and reported fluorescence quenching with neuronal death in neurodegenerative disorders (Arrasate and Finkbeiner 2005; Arrasate and others 2004; Linsley and others 2019). Fluorescence quenching can be measured serially, provided that stable microscopic access is available. Serial measurements can provide the time course of neuronal death after injury or before and after neuroprotective maneuvers. Such measures may solve the conundrums of the loss of neurons in Alzheimer disease as well as the fate of neurons in the penumbra of acute ischemic stroke.
More robust measures of neuronal death should produce fewer false-positive neurotoxic and neuroprotective results. In turn, the reduction in false-positive studies will improve the translatability of preclinical neuroprotection studies to the clinic.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: National Institutes of Health grants 1R35NS116852-01 and 5R37NS077908-08 to K.S.