
Abstract
Autism spectrum disorders (ASD) are heterogeneous neurodevelopmental conditions characterized by deficits in social communication and repetitive behaviours, collectively affecting nearly 2% of the global population. ASD is strongly heritable, with genetic variants converging on molecular pathways that regulate synaptic connectivity. Protocadherins, particularly nonclustered protocadherins (ncPCDHs), have emerged as high-confidence ASD-associated genes. By mediating cell surface adhesion and downstream signalling, ncPCDHs regulate essential neurodevelopmental processes ranging from neuronal production and migration to synaptic maturation, while generating combinatorial surface codes that organize circuit wiring—all mechanisms highly relevant to ASD. Here, we review recent findings from human genetics and rodent models, highlighting activity-dependent signalling as a central hub through which ncPCDHs coordinate synaptic refinement and maintain excitatory/inhibitory balance. Finally, we discuss how dissecting ncPCDH molecular mechanisms may clarify ASD pathogenesis and inform targeted therapeutic strategies.
Keywords
Introduction
Autism spectrum disorder (ASD) is classified by the Diagnostic and Statistical Manual of Mental Disorders (fifth edition) as a neurodevelopmental condition characterized by significant deficits in verbal and nonverbal communication and social interaction, with restrictive and repetitive patterns of behaviours, interests, or activities. Beyond these core diagnostic features, individuals with ASD frequently present with co-occurring psychiatric and medical conditions, including intellectual disability, speech delay, epilepsy, attention-deficit/hyperactivity disorder, motor control difficulties, anxiety, sleep disorders, and gastrointestinal problems (Hirota and King 2023). Individuals with autism often exhibit distinct neuroanatomic and circuit-level alterations, such as accelerated brain growth in the first years of life, regions of cortical disorganization, altered grey/white matter ratios, increased neuronal numbers, elevated excitatory synapse and spine densities, and imbalances in glutamatergic and GABAergic circuits. Collectively, these features point to widespread perturbations of fundamental neurodevelopmental programs that shape brain connectivity and function (Jiang et al 2022).
Over the past 2 decades, ASD prevalence has increased worldwide, likely due to broader diagnostic criteria and increased awareness, with current estimates of 1.5% to 2% of the population. A marked sex bias is observed, with a male:female ratio of 4.3:1, possibly reflecting hormonal or sex chromosome–linked differences (Hirota and King 2023), although underdiagnosis in females due to subtler symptom presentation and stronger social communication skills has also been proposed (Fyfe et al 2026). Despite its high prevalence and clinical impact, ASD aetiology and progression remain poorly understood, with no single factor explaining a substantial proportion of cases (Bourgeron 2015). Currently, there are no validated biomarkers for early diagnosis and no pharmacologic treatments targeting core ASD symptoms, with behavioural interventions remaining the primary therapeutic option (Hirota and King 2023).
While the relative influence of genetic and environmental factors to ASD remains debated, all epidemiologic research indicates a strong genetic component, with heritability estimated at 52% to 90% in twin and family studies (Bourgeron 2015). Advances in copy number variation analyses and in whole genome and whole exome sequencing have identified >1200 ASD-associated genes, as catalogued by the Simons Foundation Autism Research Initiative. A small subset of ASD cases presents as well-defined syndromic, typically monogenic conditions, such as fragile X syndrome (FMR1), tuberous sclerosis (TSC1/2), Rett syndrome (MECP2), Phelan-McDermid syndrome (SHANK3), and Angelman syndrome (UBE3A; Jiang et al 2022). More broadly, ASD risk is thought to arise from rare high-penetrance variants, an additive effect of common low-risk alleles, or combinations thereof, reflecting the heterogeneous genetic architecture of the disorder (Bourgeron 2015). Importantly, despite this genetic diversity, many ASD-associated genes converge on shared biological pathways—primarily synaptic development and plasticity, chromatin and transcriptional regulation, translational control, and immune responses (De Rubeis et al 2014; Jiang et al 2022).
Rodent models based on human genetic findings have proven indispensable for investigating the neurobiological mechanisms underlying ASD. Although core social deficits are intrinsically challenging to model in rodents due to their species-specific nature, a range of behavioural assays has been developed to probe these traits (Kazdoba et al 2016; Figure 1). With molecular and circuit-level analysis, these approaches have yielded critical insight into ASD-relevant pathways and serve as an important platform for preclinical therapeutic research.
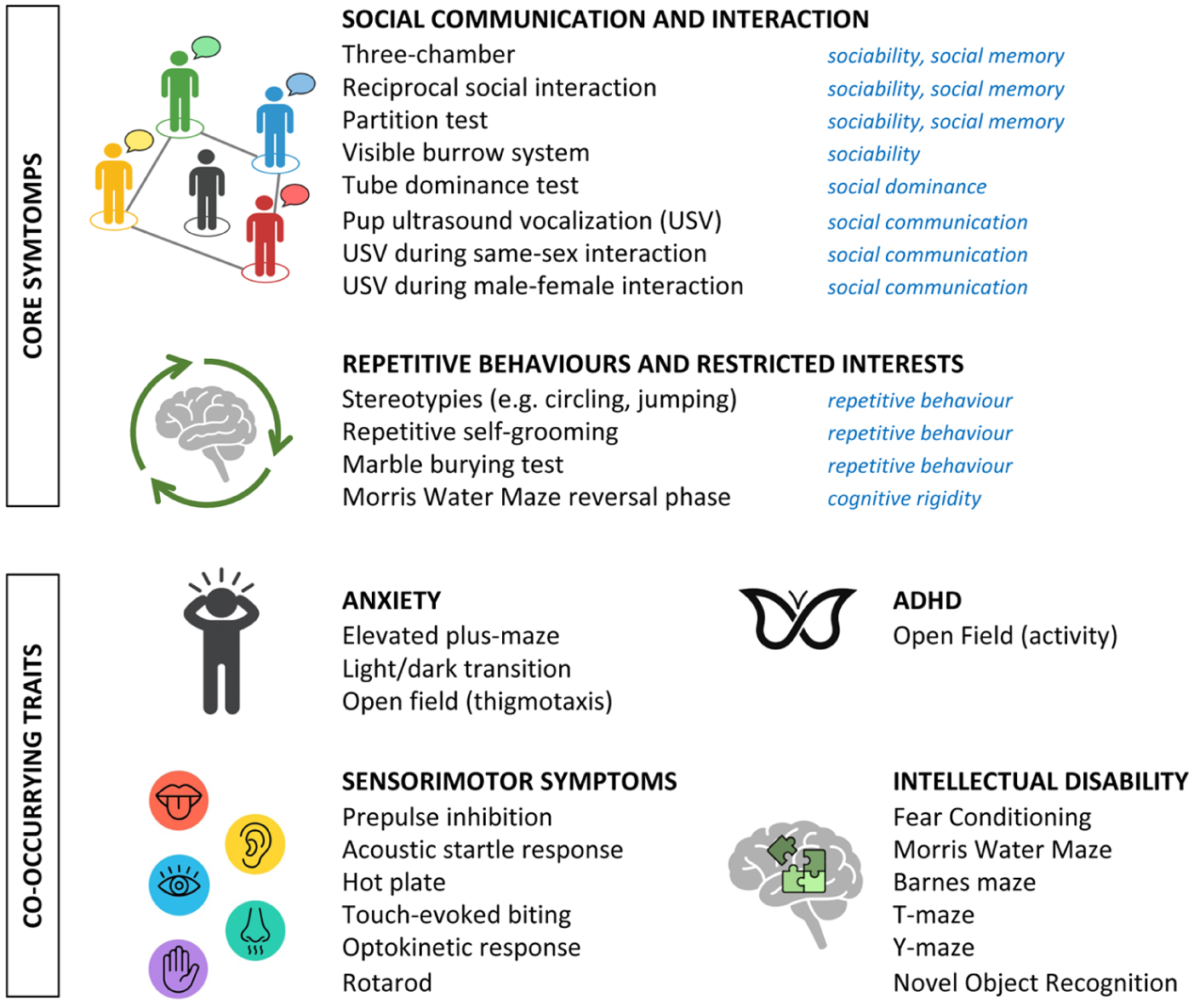
Behavioural assays used to assess core autism spectrum disorder features and co-occurring psychiatric and medical conditions in rodents. For core features (social communication and interaction; repetitive behaviours and restricted interests), the specific behavioural traits modelled by each test are indicated in blue. Detailed descriptions of each test are available elsewhere (Kazdoba et al 2016). ADHD, attention-deficit/hyperactivity disorder.
While synaptic and chromatin pathways have been extensively described elsewhere (Jiang et al 2022; Herrera et al 2024), the role of cell-cell recognition mechanisms in ASD has received less attention. Nonclustered protocadherins (ncPCDHs) provide a unique perspective in this regard: as cell adhesion molecules with tightly regulated developmental expression, they are central to establishing neuronal identity, guiding circuit formation, and organizing synapses. Growing genetic evidence implicates several family members in ASD, highlighting ncPCDHs as a key entry point for understanding disease mechanisms. Here, we integrate recent findings from human genetics and rodent models and discuss how dissecting ncPCDH regulation and mechanisms could shed light on ASD pathogenesis and inform therapeutic strategies.
Protocadherin General Features
Protocadherins (PCDHs) are single-pass transmembrane glycoproteins and constitute the largest subfamily of the cadherin superfamily, a group of calcium-dependent cell adhesion molecules involved in cell-cell recognition and tissue organization (Hirano and Takeichi 2012). Like classical cadherins, PCDHs are composed of an extracellular domain consisting of extracellular cadherin (EC) repeats, a transmembrane region, and an intracellular domain but differ in EC number and sequence composition. Based on genomic organization, PCDHs are broadly classified into clustered (cPCDHs) and nonclustered (ncPCDHs; Hulpiau and van Roy 2009).
cPCDHs, encompassing the PCDHα, β, and γ families, are encoded in tandem gene clusters within a single genomic locus. Through a remarkable combination of stochastic alternative promoter choice and alternative splicing, each cluster generates a vast repertoire of isoforms, creating extraordinary diversity in their extracellular domains while maintaining either constant intracellular regions (PCDHα and γ) or variable ones (PCDHβ; Flaherty and Maniatis 2020).
In contrast, ncPCDHs are encoded by dispersed genes across multiple chromosomes. Most ncPCDHs have been classified as members of the PCDHδ family (PCDH1, 7, 8, 9, 10, 11X, 17, 18, 19), except for PCDH12 and PCDH20, whose classification is debated (Redies et al 2005; Hulpiau and van Roy 2009; Figure 2). Based on the number of EC repeats and the amino acid sequence of the cytoplasmic domain, the PCDHδ family can be divided into 2 subgroups: PCDHδ1 (PCDH1, PCDH7, PCDH9, PCDH11X) and PCDHδ2 (PCDH8, PCDH10, PCDH17, PCDH18, PCDH19). PCDHδ has several isoforms originating from alternative splicing, which contain identical extracellular domains but differ in their cytoplasmic region. While PCDHδ2 harbours 2 conserved motifs (CM1 and CM2) in the cytoplasmic domain, PCDHδ1 has an additional conserved motif (CM3) with a putative binding site for protein phosphatase 1a (Yoshida et al 1999; Vanhalst et al 2005), whereas conserved motifs are absent in PCDH12 and PCDH20.
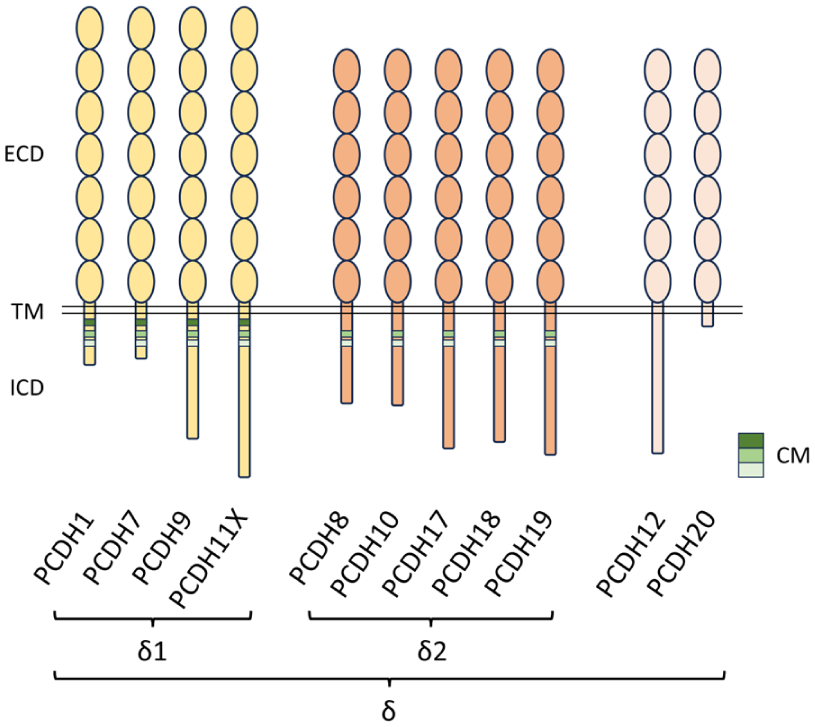
Molecular structure of ncPCDH family members. ncPCDHs contain an N-terminal ECD composed of conserved EC repeats (ellipses), which show low sequence similarity to those of classical cadherins, a single TM domain, and a C-terminal ICD. ncPCDHs include the PCDHδ group, subdivided into δ1 (7 EC repeats) and δ2 (6 EC repeats) subgroups. Within their intracellular domains, δ1 PCDHs and δ2 PCDHs contain 3 and 2 CMs, respectively, whereas PCDH12 and PCDH20 lack CM motifs. CM, conserved motif; EC, extracellular cadherin; ECD, extracellular domain; ICD, intracellular domain; ncPCDH, nonclustered protocadherin; PCDH, protocadherin; TM, transmembrane.
cPCDHs engage in homophilic trans interaction via EC1–EC4 domains in a head-to-tail orientation (Goodman et al 2016), mediating cell-cell recognition and adhesion. In addition, cPCDH can form homophilic and heterophilic cis interactions through their extracellular domains. These cis associations expand molecular diversity by generating novel trans-binding specificities, creating a highly diverse cell surface recognition code that contributes to neuronal identity (Flaherty and Maniatis 2020). The simultaneous engagement of cis and trans interactions allows cPCDH to assemble into characteristic higher-order zipper-like structures. Depending on cellular context, the interplay between these interactions can promote either adhesion or repulsion and is thought to underlie the critical role of cPCDHs in self-discrimination (Pancho et al 2020).
Similar to cPCDHs, ncPCDHs can engage in homophilic trans interactions (Cooper et al 2016; Harrison et al 2020), and cis interactions across ncPCDHs have been reported (Pederick et al 2018; Mincheva-Tasheva et al 2024). However, cis associations do not appear to be mediated by the extracellular domain, and available evidence suggests that ncPCDHs do not form zipper-like assemblies (Harrison et al 2020). As observed for cPCDHs, combinatorial expression of δ-PCDHs supports self-recognition such that cells expressing identical δ-PCDH preferentially associate (Hayashi et al 2017; Pederick et al 2018; Mincheva-Tasheva et al 2024). Beyond contributing to cell surface diversity, ncPCDHs display larger and distinctive variety in their cytosolic domains as compared with cPCDHs, likely enabling intracellular interactions with diverse partners and supporting specialized signalling roles.
PCDHs are broadly present throughout the central nervous system, including the cortex, hippocampus, amygdala, cerebellum, thalamus, hypothalamus, basal ganglia, limbic structures, and sensory systems. At the same time, their expression is tightly spatiotemporally regulated. Many PCDHs display dynamic developmental expression profiles (S-Y Kim et al 2007; Bassani et al 2018; Miozzo et al 2024), often peaking during postnatal stages, consistent with roles in synaptogenesis. Within individual brain regions, ncPCDHs frequently exhibit spatially restricted patterns. These include layer- and region-specific localization in the cortex (S-Y Kim et al 2007; Krishna-K et al 2011) and selective expression in defined amygdala subdivisions (Hertel et al 2012), supporting combinatorial codes that contribute to neuronal population specification and circuit architecture. Accordingly, PCDHs have been implicated in a large range of neurodevelopmental processes, including neural progenitor proliferation, cell recognition, and neuronal migration, as well as synapse formation, maturation, and plasticity (Peek et al 2017). Consistent with these roles, genetic alterations or dysfunction in PCDH genes has been widely linked to neurodevelopmental and psychiatric disorders (Mancini et al 2020).
While the involvement of cPCDHs in brain conditions has been extensively covered by dedicated reviews (Flaherty and Maniatis 2020; Jia and Wu 2020), ncPCDHs have received less attention. Yet human genetic studies have repeatedly identified ncPCDH genes (PCDH8, PCDH9, PCDH10, PCDH11X, PCDH19) as risk factors across syndromic and nonsyndromic ASD. In the following sections, we review ASD-linked ncPCDHs individually, summarizing genetic evidence and recent insights from mouse lines, to examine how their dysfunction affects behaviours (Supplemental Table 1) and neurodevelopmental processes (Supplemental Table 2) relevant to ASD. Despite their misleading nomenclature, adhesion proteins such as PCDH15, PCDH21, and PCDH23 are generally not classified as ncPCDHs because they are phylogenetically distinct from this group and lack its characteristic structural features. Consequently, they are not discussed in the present review.
PCDH19
Pcdh19 and Developmental and Epileptic Encephalopathy 9
PCDH19 first attracted widespread attention in 2008, when pathogenic variants were recognized as the cause of developmental and epileptic encephalopathy 9 (DEE9; Dibbens et al 2008; Depienne et al 2009). DEE9 is a convulsive disorder limited to females and characterized by early-onset seizures that commonly remit during adolescence and are frequently drug resistant (Samanta 2020). Affected individuals often present with a wide spectrum of neurodevelopmental and neuropsychiatric symptoms, with intellectual disability and autistic features among the most common (Smith et al 2018; Kolc et al 2019; Samanta 2020).
More than 175 PCDH19 variants have been reported, most being missense point mutations in the extracellular domain that are thought to cause loss of function (Niazi et al 2019). Remarkably, PCDH19 mutations cause DEE9 almost exclusively in heterozygous females. As the PCDH19 gene is located on the X chromosome, random X chromosome inactivation in females leads to a mosaic brain composed of neurons expressing either the wild type or mutant allele. This unusual inheritance pattern, reminiscent of the mechanism first proposed for Ephrin B1–related craniofrontonasal syndrome (Wieland et al 2004), led to the cellular interference hypothesis, which proposes that aberrant interactions between these 2 neuronal populations perturb PCDH19-mediated cell-cell recognition, resulting in impaired neuronal communication and network assembly (Depienne et al 2009). Supporting this model, DEE9 has been reported in males with somatic mosaicism, as well as in individuals with Klinefelter syndrome (XXY), implicating mosaic expression as a key pathogenic mechanism (Mancini et al 2020).
Genetic mouse models have reinforced the notion that Pcdh19 mosaicism produces unique neurobiological consequences not observed in nonmosaic contexts. At least 3 independent laboratories demonstrated that mosaic Pcdh19 expression in heterozygous females drives striking segregation of PCDH19+ and PCDH19- cortical neurons (Hayashi et al 2017; Pederick et al 2018; J Lim et al 2019), accompanied by abnormal cortical network activity (Pederick et al 2018). Crucially, deletion of both Pcdh19 alleles does not cause these defects, supporting the cellular interference model (Pederick et al 2018). Abnormal clustering of PCDH19+ and PCDH19- neurons has also been recently confirmed in human cortical organoids (Niu et al 2024). At the synaptic level, heterozygote females from a distinct Pcdh19 mosaic line, but not hemizygote males, display defects in mossy fiber synaptic structure, physiology, and related cognitive behaviours (Hoshina et al 2021). These phenotypes arise from a mismatch of PCDH19 availability in pre- and postsynaptic compartments (Hoshina et al 2021; Mincheva-Tasheva et al 2021), implicating mosaic Pcdh19 expression as a pathogenic driver.
Although Pcdh19 mutants do not display overt brain malformations and spontaneous seizures, multiple mosaic models show increased susceptibility to induced seizures (Bassani et al 2018; Cwetsch et al 2022; Robens et al 2022; Giansante et al 2023) and moderate cognitive deficits (Hayashi et al 2017; Hoshina et al 2021; Cwetsch et al 2022; Giansante et al 2023). However, another study examining heterozygous and homozygous Pcdh19 knockout (KO) females reported increased susceptibility to induced seizures in both genotypes as compared with wild type animals, whereas hemizygote males were unaffected (Rakotomamonjy et al 2020). While these findings reinforce the link between Pcdh19 and seizure vulnerability in females, they also challenge the cellular interference hypothesis as the sole mechanism underlying DEE9 manifestations.
Pcdh19 and ASD
Large clinical evidence supports a contribution of PCDH19 mutations to ASD. Autistic features are common in females heterozygous for PCDH19 mutations diagnosed with DEE9 (Scheffer et al 2008; Smith et al 2018; Kolc et al 2019; Samanta 2020) and in rare mosaic males with DEE9-like phenotypes (Depienne et al 2009; de Lange et al 2017; Parmeggiani et al 2024). Importantly, ASD has also been documented in hemizygous males carrying PCDH19 mutations in the absence of DEE9 (van Harssel et al 2013; Chouery et al 2023) and in unbiased genetic screening of ASD cohorts (Piton et al 2011; Zhou et al 2022; Mendes et al 2025; Suspitsin et al 2025). These observations suggest that ASD associated with PCDH19 mutations may not primarily depend on cellular interference mechanisms and that altered PCDH19 dosage alone may be sufficient to disrupt neurodevelopmental pathways relevant to ASD.
Mouse studies seem to corroborate this view. Pcdh19 hemizygous males lacking Pcdh19 expression display reduced sociability in the 3-chamber test and the reciprocal interaction assay, with increased repetitive behaviours. Pcdh19 heterozygote females also displayed markedly reduced sociability (J Lim et al 2019). Furthermore, mosaic females from an independent Pcdh19 line showed increased repetitive behaviour (Giansante et al 2023). In rats, local mosaics of Pcdh19-downregulated and control cells in the somatosensory cortex impair early social behaviours, reducing pups huddling and ultrasonic vocalization (USV), and lead to poor sociability in adulthood (Cwetsch et al 2022). However, 2 other mouse studies reported no social deficits in Pcdh19 mutants (Hayashi et al 2017; Galindo-Riera et al 2021). Beyond social traits, multiple laboratories reported moderate cognitive deficits and sensorimotor alterations across Pcdh19 mutants (Hayashi et al 2017; Hoshina et al 2021; Cwetsch et al 2022; Giansante et al 2023).
Taken together, loss-of-function manipulation of Pcdh19 demonstrates potential to model key ASD-relevant behaviours, including social interactions, social communication, and repetitive behaviours. These effects do not seem to be restricted to females, suggesting that Pcdh19 may contribute to ASD independently of cellular interference mechanisms, consistent with reports of male patients with autism carrying PCDH19 mutations without DEE9 (Piton et al 2011; van Harssel et al 2013; Chouery et al 2023; Mendes et al 2025; Suspitsin et al 2025). Nonetheless, discrepancies across studies remain, likely reflecting differences in mutant lines and behavioural paradigms. Systematic investigation using standardized assays across Pcdh19 models would help clarify the contribution of Pcdh19 loss of function to ASD-related traits, the relative impact of mosaic vs uniform protein loss, and potential sex-specific effects.
Pcdh19 Neuronal and Molecular Mechanisms
Research in rodents has established Pcdh19 as a central regulator of neurodevelopment and brain function. Pcdh19 is expressed in the brain from embryonic stages (S-Y Kim et al 2007; Hayashi et al 2017; Bassani et al 2018), where it is enriched in neural progenitors and regulates cell polarity and neurogenesis, as shown in cultures, organoids (Homan et al 2018; Borghi et al 2021; Niu et al 2024), and in vivo (Cooper et al 2015; Fujitani et al 2017; Lv et al 2019; Biswas et al 2021). Pcdh19 expression peaks during the early postnatal period, particularly in limbic regions such as the amygdala, hippocampal CA1 and CA3 fields, subiculum, and selected hypothalamic areas—circuits strongly implicated in seizure generation (S-Y Kim et al 2007; Pederick et al 2016; Hayashi et al 2017; Bassani et al 2018; Schaarschuch and Hertel 2018; Cwetsch et al 2022). This temporal and regional expression profile is also consistent with demonstrated roles of Pcdh19 in pyramidal neuron and cortical interneuron migration (Bassani et al 2018; Cwetsch et al 2022; Pancho et al 2022), and neuronal clustering (Hayashi et al 2017; Pederick et al 2018), as well as hippocampal synapse development, activity, and plasticity (Bassani et al 2018; Hoshina et al 2021; Giansante et al 2023).
At the molecular level, the contribution of PCDH19 to neurodevelopmental and synaptic processes likely relies on cell-cell recognition and adhesion mechanisms through homophilic trans interactions (Tai et al 2010; Emond et al 2011; Hayashi et al 2017; Bisogni et al 2018; Pederick et al 2018; Harrison et al 2020) and cis interactions with N-cadherin and other PCDHs (Biswas et al 2010; Emond et al 2011; Pederick et al 2018; Hoshina et al 2021). PCDH19 also associates with key regulators of actin and microtubule dynamics, including the WASP family verprolin homologous protein (WAVE) complex and Rho GTPase regulators (Tai et al 2010; Chen et al 2014; de Nys et al 2024), supporting a role in adhesion junction formation and cytoskeleton remodelling. PCDH19 was shown to localize to synapses in hippocampal mossy fibers (Hoshina et al 2021) and in neuronal cultures upon overexpression (Hayashi et al 2017). However, endogenous PCDH19 was detected only in a subset of synapses in cultured neurons, suggesting that it might not represent a constitutive synaptic component (Hayashi et al 2017; Mincheva-Tasheva et al 2021). At the synapse, PCDH19 was shown to regulate GABAergic transmission via direct interaction with the GABAA receptor in primary neurons (Bassani et al 2018; Serratto et al 2020). In addition, PCDH19 has been implicated in transcriptional and epigenetic regulation through interplay with Wnt/β-catenin signalling, the NONO–estrogen receptor alpha axis, and the histone demethylase LSD1 (Pham et al 2017; Biswas et al 2021; Gerosa et al 2022; de Nys et al 2024).
Taken together, Pcdh19 functions in neuronal generation, migration, clustering, and synaptic development likely underlie the profound alterations in neuronal excitability, circuit wiring, and network activity observed in Pcdh19 models (Pederick et al 2018; Mincheva-Tasheva et al 2021; Giansante et al 2023) and may contribute to some of the structural and functional abnormalities reported in patients with DEE9 and ASD.
PCDH9
Strong genetic evidence implicates PCDH9 in ASD. Associations between PCDH9 and ASD were first reported through the identification of de novo and inherited copy number variations in multiple cohorts (Marshall et al 2008; Bucan et al 2009; Girirajan et al 2013). These findings were replicated using independent statistical approaches (Yang et al 2022) and were recently corroborated in a cohort of Korean patients with autism (S-W Kim et al 2024). In addition, reduced PCDH9 mRNA levels were detected in lymphoblasts from patients with ASD (R Luo et al 2012). Beyond ASD, a meta-analysis of 3 genome-wide association studies identified a single-nucleotide polymorphism in PCDH9 as a risk locus for major depressive disorder and cognitive impairment (Xiao et al 2018).
Pcdh9 is broadly expressed in the CNS, including the olfactory bulb, hippocampus, striatum, cortex, and cerebellum, from early developmental stages through adulthood (Asahina et al 2012; Miozzo et al 2024). Extensive behavioural characterization of Pcdh9-null mice has revealed alterations in specific traits relevant to ASD (Bruining et al 2015). While overall sociability is preserved, these animals exhibit consistent impairments in long-term social recognition, as independently confirmed by another group (Miozzo et al 2024). In the cognitive domain, Pcdh9-deficient animals show reproducible deficits in the novel object recognition task (Bruining et al 2015; Miozzo et al 2024), whereas performance in other memory assays remained largely unaffected. Remarkably, Pcdh9 KO mice display multiple defects in sensory perception and sensorimotor competence, as evidenced by reduced touch-evoked biting, deficits in prepulse inhibition, and decreased latency to fall in the rotarod test (Bruining et al 2015). Experiments on a distinct Pcdh9 mutant line also uncovered impairments in the optokinetic response (Uemura et al 2022). These behavioural phenotypes are accompanied by structural and functional alterations in specific brain regions. Pcdh9 KO mice exhibit reduced cortical thickness and neuronal counts in the deep layers of the somatosensory cortex, along with reduced dendritic arborization and increased spine density in pyramidal neurons, possibly at the origin of the somatosensory processing dysfunctions (Bruining et al 2015).
More recently, new studies have shed light on the neuronal and molecular functions of Pcdh9. PCDH9 is predominantly localized at glutamatergic synapses in primary neurons, with hippocampal expression peaking during the first postnatal week, a critical period for synaptogenesis (Miozzo et al 2024). Ultrastructural analyses of Pcdh9 KO mice revealed enlarged presynaptic terminals and postsynaptic densities in neurons of the CA1 region. This synaptic overgrowth is supported by moderate but broad transcriptional upregulation of synaptic genes and aberrant levels of the SHANK2/CORTACTIN pathway involved in synapse morphogenesis. At the electrophysiologic level, these structural and molecular changes are accompanied by enhanced miniature excitatory postsynaptic currents and reduced network activity, highlighting a critical role for Pcdh9 in shaping excitatory synapse morphology and function in the CA1 (Miozzo et al 2024). Another study from the same group implicated PCDH9 in neuronal intracellular signalling (Miozzo et al 2026). PCDH9 undergoes activity-dependent cleavage by matrix metalloproteases in primary neurons, generating a C-terminal fragment (CTF) that can translocate to the nucleus. PCDH9 CTF overexpression leads to increased dendritic length, spine density, and excitatory synaptic transmission, revealing an unexpected function for PCDH9 in the nucleus. These findings identify PCDH9 as a novel activity-dependent synaptonuclear messenger involved in synaptic plasticity, with potential relevance to ASD.
Collectively, mouse studies highlight Pcdh9 as an important player in cortical and hippocampal neurodevelopment, with relevant consequences on sensory processing, social recognition, and cognitive functions, thereby strengthening its link with ASD and psychiatric disorders.
PCDH10
Multiple independent genomic approaches, including copy number variation analyses, studies in consanguineous populations, and exome sequencing, have consistently implicated PCDH10 as an ASD-susceptible gene (Morrow et al 2008; Bucan et al 2009; Takata et al 2018). Furthermore, PCDH10 has been linked to conditions comorbid with ASD, including obsessive-compulsive disorder, where PCDH10 variants modulate selective serotonin reuptake inhibitor treatment response (Qin et al 2016), and Tourette syndrome (Depienne et al 2019). Beyond ASD, PCDH10 has been associated with major depressive disorder (Roberson-Nay et al 2020) and identified as a susceptibility gene for schizophrenia and bipolar disorder (Tang et al 2019).
Pcdh10 is widely expressed across the brain and especially enriched in the olfactory and visual systems and in limbic structures including the amygdala (S-Y Kim et al 2007; Uemura et al 2007; Aerts et al 2024), a brain area critically involved in social behaviour and ASD. The role of Pcdh10 in mouse brain and behaviour is debated. Early studies on a Pcdh10 KO line obtained by replacing the first exon with a LacZ-Neo cassette found severe defects in striatal and thalamocortical projections in heterozygous animals (Pcdh10LacZ-Neo/+; Uemura et al 2007). Behavioural characterization revealed multiple alterations in social traits. Pups of both sexes exhibit increased USV, a feature observed in other ASD models and suggestive of altered social communication. Adult males display reduced sociability in the 3-chamber test. This phenotype is rescued by the NMDAR agonist
In contrast, a group using a Crispr/Cas9-generated Pcdh10 KO line without a LacZ-Neo cassette failed to reproduce these findings and suggested that they might be artifacts from the cassette insertion (Hoshina et al 2022). These Pcdh10 KO mice exhibit normal axon projections, spine density, and spine morphology in the basolateral amygdala but show impaired development of excitatory synapses in pre- and postsynaptic compartments. Male mutants present intact sociability, mildly reduced social recognition, and normal USV (Hoshina et al 2022).
However, a recent study using a Cre-loxP strategy to ubiquitously delete Pcdh10 reproduced the increased USV emission initially reported by Schoch et al 2017. The authors confirmed the same phenotype following selective Pcdh10 deletion in Gsh2-lineage interneurons, which was accompanied by substantial reduction of this cell population in the basolateral amygdala, thus providing a putative neuronal mechanism underlying the USV phenotype (Aerts et al 2024).
Despite the aforementioned discrepancies, convergent evidence supports a contribution for Pcdh10 in ASD-relevant circuitry of the basolateral amygdala. Schoch et al 2017 and Hoshina et al 2022 implicated Pcdh10 in glutamatergic synapse development, while Aerts et al 2024 demonstrated its requirement for interneuron maintenance in this region. While further studies are needed to clarify the contribution of Pcdh10 to sociability and social recognition, multiple Pcdh10 mutant lines consistently display pup USV abnormalities, a proxy for early-life communication deficits and anxiety-related behaviours related to ASD. Overall, Pcdh10 mutants retain potential to capture biologically meaningful aspects of social dysfunctions in ASD.
PCDH8
Although genetic evidence linking PCDH8 to brain disorders is more limited than for other PCDHs, whole exome sequencing studies in women who are autistic have identified PCDH8 as an ASD-associated gene (Butler et al 2015). The function of PCDH8 in the nervous system remains poorly understood, with current knowledge derived mainly from in vitro and developmental studies rather than behavioural analyses. Pcdh8 is broadly expressed throughout the rodent brain, with higher levels in the olfactory bulb, superficial cortical layers, and hippocampus (S-Y Kim et al 2007). Recent work shows that Pcdh8 regulates cortical development by controlling cell proliferation and neuronal fate through interplay with the transcription factor Dbx1 (Cwetsch et al 2026). Beyond development, Pcdh8 is robustly induced by neuronal activity, localizes to synapses, and can modulate synaptic function. Antibody-mediated blockade of PCDH8 reduces miniature excitatory postsynaptic current amplitude and impairs long-term potentiation (LTP) in hippocampal slices, supporting an active role in activity-dependent synaptic plasticity (Yamagata et al 1999). At the same time, cultured Pcdh8−/− hippocampal neurons exhibit increased dendritic spine density, suggesting that Pcdh8 negatively regulates spine formation. Mechanistically, PCDH8 interacts with N-cadherin and triggers its endocytosis via TAO2β and p38 MAPK signalling, a pathway proposed to function as a delayed, activity-dependent homeostatic mechanism to constrain synaptic growth and stabilize networks (Yasuda et al 2007). Given the central involvement of activity-dependent synaptic processes in ASD neuropathology (Yap and Greenberg 2018; Faust et al 2021), these findings encourage further investigation. Phenotypic characterization of Pcdh8−/− mice across ASD-relevant behavioural domains will be essential to establish translational relevance.
PCDH11X
Over the past decade, PCDH11X has emerged as an ASD risk gene across multiple genetic studies, a finding of particular interest given its location on the X chromosome, consistent with the higher prevalence of the disorder in males. The first direct link between PCDH11X and ASD was the discovery of a hemizygous splice-site variant in a male with ASD (ET Lim et al 2013), later supported by genomic studies identifying loss-of-function variants in ASD families (C Yuen et al 2017). More recently, PCDH11X was confirmed as an ASD candidate gene in an integrated analysis of de novo and inherited coding variants in >40,000 ASD cases (Zhou et al 2022) and independently implicated in additional sequencing screens (Miyake et al 2024). Pathogenic variants have also been associated with intellectual disability and severe language impairment, including developmental dyslexia (Veerappa et al 2013). Given that communication deficits represent a core ASD dimension, altogether these findings position PCDH11X as a strong candidate gene for ASD-relevant neurodevelopmental pathways.
However, knowledge of the role of Pcdh11x in neurodevelopment and brain integrity remains limited. Pcdh11x is expressed in various brain areas, including specific regions and layers of the cortex, hippocampus, and ventricular/subventricular zones (S-Y Kim et al 2007). Early studies demonstrated that Pcdh11x regulates neural stem cell (NSC) proliferation and differentiation in vivo (Zhang et al 2014) and negatively controls dendrite branching via PI3K/AKT signalling in cultured cortical neurons (Wu et al 2015). More recent work implicates Pcdh11x in adult brain rewiring: its disruption in granule cells alters mossy fiber synapse targeting, with new synapses frequently formed on cell soma in addition to dendrites (W Luo et al 2022). In vivo behavioural assessment of Pcdh11x mutants will be crucial to determine how these cellular and circuit-level alterations contribute to ASD-relevant phenotypes.
ncPCDHs in ASD-Related Neurodevelopmental Mechanisms
In the following section, we evaluate how dysfunction of ASD-linked ncPCDHs converges on the major interconnected pathways involved in ASD (Figure 3). These frameworks are not mutually exclusive but instead represent complementary levels at which ncPCDHs may shape ASD-relevant neurobiology.
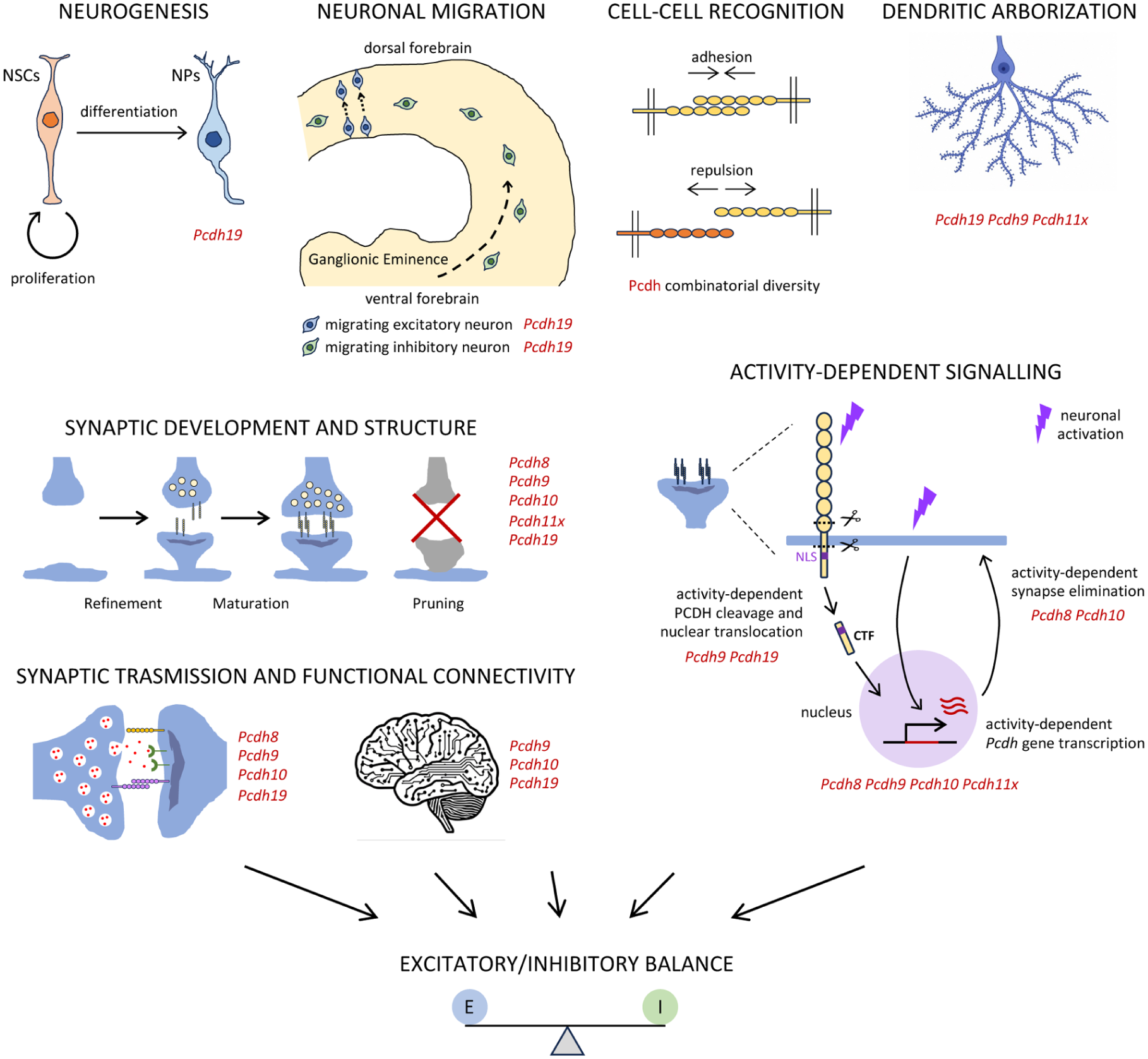
Neurodevelopmental processes relevant to ASD and associated nonclustered PCDH family members. Schematic representation of key neurodevelopmental processes and mechanisms implicated in ASD, including neurogenesis, neuronal migration, cell-cell recognition, dendritic arborisation, synaptic development, synaptic transmission and functional connectivity, and activity-dependent signalling—all able to influence the excitatory/inhibitory balance. For each process, ASD-linked Pcdh genes with reported functional involvement are listed alongside. The figure summarises experimental evidence from rodent models and in vitro systems. ASD, autism spectrum disorder; NPs, neuronal precursors; NSCs, neural stem cells; PCDH, protocadherin.
ncPCDHs in Neurogenesis and Early Brain Overgrowth
Approximately 20% of children with ASD present with macrocephaly, and converging imaging studies support a model of accelerated brain growth during precocious childhood. Although the underlying cellular mechanisms remain debated, dysregulated neurogenesis and neuronal outgrowth have been proposed as a contributing factor for ASD pathophysiology (Currey et al 2025).
In this context, Pcdh19 has been implicated in the control of neural progenitor proliferation and differentiation across multiple experimental systems (Cooper et al 2015; Fujitani et al 2017; Homan et al 2018; Lv et al 2019; Biswas et al 2021; Borghi et al 2021). Interestingly, in zebrafish neuroepithelium, multiple ncPCDHs have been shown to regulate neural progenitor proliferations by modulating Wnt/β-catenin signalling (Biswas et al 2021), a pathway strongly linked to ASD (Jiang et al 2022). In cancer, PCDH loss of function is frequently associated with enhanced cell division through dysregulation of Wnt/β-catenin and PI3K/AKT/mTOR signalling (Mah and Weiner 2017; Pancho et al 2020), another pathway implicated in ASD pathophysiology (Jiang et al 2022). Whether analogous mechanisms operate during mammalian brain formation remains largely unexplored, but these observations suggest a potential link between PCDH and early neurodevelopmental trajectories in ASD pathogenesis.
ncPCDHs in Dendritic Spine Development and Synapse Organization
Robust evidence from human studies and animal models has led to the conceptualization of ASD as a disorder of synaptic development. A large fraction of ASD-associated genes encodes proteins involved in synapse organization and function, including synaptic scaffolds, neurotransmitter receptors, ion channels, cell adhesion molecules, and cytoskeletal regulators (De Rubeis et al 2014; Bourgeron 2015). Consistent with this genetic landscape, postmortem ASD brains show increased cortical dendritic spine density with transcriptional signatures of impaired synaptic pruning, suggesting an underpruning phenotype that is recapitulated across multiple animal models (Faust et al 2021; Jiang et al 2022).
Within this framework, ncPCDHs have emerged as important modulators of synapse and dendritic spine density. Studies across several ncPCDH mutants revealed alterations in excitatory synapse number, with effects that vary in direction depending on the specific family member and brain region examined. Increased spine density has been reported following loss of Pcdh10 (Uemura et al 2007) and Pcdh8 (Yasuda et al 2007), in agreement with their proposed role in synapse elimination, as well as after Pcdh9 deletion (Bruining et al 2015). On the contrary, reduced synapse number has been observed in models of mosaic Pcdh19 expression (Mincheva-Tasheva et al 2021; Giansante et al 2023). Beyond spine density, multiple ncPCDHs have been implicated in the regulation of synaptic architecture. In the hippocampus, manipulation of Pcdh19 alters excitatory and inhibitory (E/I) synapse organization (Bassani et al 2018; Hoshina et al 2021; Giansante et al 2023), and Pcdh9 deletion leads to aberrantly enlarged synapses (Miozzo et al 2024), whereas Pcdh10 influences synaptic structure in the amygdala (Schoch et al 2017; Hoshina et al 2022).
Mechanistically, ncPCDHs may regulate synaptic stability, turnover, and organization through interactions mediated by distinct domains, including intracellular binding to postsynaptic density scaffolding proteins (eg, PCDH10-PSD95; Tsai et al 2012) and cis association with other cell adhesion molecules, such as PCDH8 and PCDH19 interaction with N-cadherin (Yasuda et al 2007; Biswas et al 2010; Emond et al 2011; Hoshina et al 2021). ncPCDHs emerge therefore as critical and versatile players in synaptic development, while important questions remain regarding the molecular mechanisms and context dependence of their actions.
ncPCDHs in the E/I Balance
An imbalance in E/I neuronal signalling has long been proposed as a core mechanism underlying ASD, with converging evidence pointing to prominent GABAergic dysfunction (Sohal and Rubenstein 2019). Loss-of-function alterations in GABA-related pathways are well documented in postmortem and in vivo clinical studies, and preclinical models consistently report deficits in the GABAergic system. E/I balance is regulated across multiple hierarchical levels, from cellular composition and synapse development to synaptic function, circuit organization, and network dynamics (Lee et al 2017). Accumulating evidence indicates that ncPCDHs operate across most, if not all, of these layers.
At the cellular level, ncPCDHs influence neuronal number and positioning, thereby shaping the substrate upon which E/I balance is established. Beyond regulating neural progenitor proliferation and differentiation, Pcdh19 controls neuronal migration in the cortex and hippocampus (Pederick et al 2016; Bassani et al 2018; Cwetsch et al 2022), all processes essential for appropriate E/I cell distribution and circuit wiring. Although ncPCDH functions in interneurons remain largely understudied, alterations in Pcdh19 affect interneuron migration (Pancho et al 2022), and selective ablation of Pcdh10 in a defined interneuron population reduces its abundance in the basolateral amygdala (Aerts et al 2024), directly affecting inhibitory circuit composition.
More broadly, ncPCDHs contribute to a combinatorial molecular code that specifies cell-cell interactions and wiring specificity. The number and combination of ncPCDHs expressed at the cell surface determine interaction selectivity, modulated by interactions with cPCDHs and other cell adhesion molecules (Bisogni et al 2018; Mincheva-Tasheva et al 2024). Disruption of this code leads to abnormal cell segregation in Pcdh19 mosaics (ET Lim et al 2013; Hayashi et al 2017; Pederick et al 2018; Niu et al 2024) and impaired cortical microcircuit organization (Lv et al 2019).
Regarding neuronal morphology and connectivity, numerous studies have shown that ncPCDHs control dendritic arborization (Bruining et al 2015; Wu et al 2015; Bassani et al 2018; Ishii et al 2019; Mincheva-Tasheva et al 2021). ncPCDHs have also been implicated in multiple aspects of axonal development across vertebrate systems. They regulate retinal axon elongation and pathfinding in Xenopus (Piper et al 2008; Leung et al 2013) and control axonal arborization in zebrafish (Biswas et al 2014). Whether ncPCDHs play a broad role in axonal extension in mammals, however, remains debated. While some studies have implicated Pcdh10, Pcdh19, and Pcdh17 in axonal projection development (Uemura et al 2007; Hayashi et al 2014; Mincheva-Tasheva et al 2021), other groups reported no observable effects on axon growth and targeting following disruption of these genes (Hoshina et al 2013; Hayashi et al 2017; Galindo-Riera et al 2021; Hoshina et al 2022). Beyond axon extension itself, specific ncPCDHs have been linked to later stages of circuit assembly and refinement. Pcdh19 mutations have been associated with defects in olfactory sensory neuron coalescence during postnatal development and regeneration (Martinez et al 2023), whereas Pcdh11x has been implicated in mossy fiber target specification during sprouting (W Luo et al 2022).
Structural and synaptic alterations in ncPCDH mutants are often accompanied by defects in neurotransmission, potentially affecting the E/I balance. Pcdh19 mosaic animals display impairments in synaptic transmission and plasticity at CA1 and mossy fiber synapses (Hoshina et al 2021; Giansante et al 2023), and effects on synaptic functions have been reported for Pcdh9 and Pcdh8 in the hippocampus (Yamagata et al 1999; Miozzo et al 2024) and for Pcdh10 in the amygdala (Hoshina et al 2022). Importantly, Pcdh19 has been directly implicated in GABAergic transmission, a crucial component of E/I balance. PCDH19 physically interacts with GABAA receptors, promoting their surface availability and modulating channel gating, thereby regulating inhibitory currents and contributing to hyperexcitability when disrupted (Bassani et al 2018; Serratto et al 2020).
At the circuit and network level, ncPCDH alterations result in complex and sometimes paradoxical effects on neuronal activity. In a Pcdh19 mosaic model, functional hyperconnectivity within limbic circuits was observed, a feature also reported in patients with and models based on ASD, whereas the E/I ratio was reduced despite single-neuron hyperexcitability (Giansante et al 2023). Similarly, Pcdh9 deletion increases miniature excitatory postsynaptic current frequency in dCA1 pyramidal neurons yet reduces overall hippocampal network activity and synchronization (Miozzo et al 2024). Both these examples highlight how multilevel disruptions in migration, wiring, and synaptic maturation can converge on emergent circuit dysfunction. In Pcdh10 haploinsufficient mice, gamma-frequency transmission is altered, consistent with the documented association between gamma-band abnormalities and ASD. Importantly, these deficits are partially rescued by the GABABR agonist baclofen, a promising compound for ASD treatment, underscoring the translational relevance of this phenotype (Port et al 2017; Schoch et al 2017).
Together, these findings position ncPCDHs as critical regulators of E/I balance, influencing neuronal composition, wiring specificity, synaptic function, and emergent network dynamics. Alteration of these developmental processes may contribute to circuit-level dysregulation characteristic of ASD.
ncPCDHs in Activity-Dependent Signalling
Activity-dependent signalling networks are recognized as central pathways disrupted in ASD. Neuronal activity triggers local synaptic changes, modulates translation, and activates transcriptional programs that direct synaptic plasticity and function. This is particularly critical during the first years of life, when sensory experience guides synapse refinement, maturation, and pruning, thereby shaping the E/I balance. Many core ASD features emerge during this period, suggesting that disruption of experience-dependent synaptic development might contribute to the disorder (Faust et al 2021). Numerous ASD-associated genes consistently regulate and are regulated by activity-dependent processes, highlighting these networks as key mediators of neurodevelopmental vulnerability (Yap and Greenberg 2018).
A rapidly expanding body of evidence links ncPCDH to activity-dependent signalling. Pcdh10 provides one of the most compelling examples. Pcdh10 expression is cooperatively regulated by the transcription factor MEF2 and the translational regulator FMRP, both activity-responsive factors encoded by syndromic ASD genes. Functionally, PCDH10 interacts with PSD-95 and promotes its targeting to the proteasome, thereby facilitating activity-dependent synapse elimination (Tsai et al 2012), a process strongly implicated in ASD. Earlier work similarly identified Pcdh8 as an activity-induced gene that negatively regulates dendritic spine formation (Yamagata et al 1999; Yasuda et al 2007). More broadly, transcriptional profiling of hippocampal excitatory neurons identified multiple additional ncPCDHs as activity-induced genes, including Pcdh9, Pcdh11x, and Pcdh17 (Fernandez-Albert et al 2019).
Beyond being induced by neuronal activity, recent studies indicate that PCDHs can directly participate in activity-dependent transcriptional regulation through proteolytic processing and translocation to the nucleus. PCDH19 undergoes activity-dependent cleavage initiated by the matrix metalloprotease ADAM10, generating a CTF that migrates to the nucleus. There, PCDH19 CTF associates with the chromatin modifier LSD1 and suppresses the expression of immediate-early genes, potentially acting as a homeostatic brake to limit excessive neuronal excitation (Gerosa et al 2022). Similarly, PCDH9 displays activity-dependent matrix metalloprotease–mediated cleavage in cultured neurons, producing a nuclear CTF that modulates neuronal morphology and function, likely via transcriptional regulation (Miozzo et al 2026). If confirmed in the brain, such a mechanism could account for the dysregulation of synaptic gene expression observed in Pcdh9 KO hippocampal neurons (Miozzo et al 2024). Although these nuclear pathways remain to be fully validated in vivo, available evidence suggests that PCDH9 and PCDH19 nuclear signalling may serve as a negative feedback mechanism that fine-tunes activity-dependent gene expression.
In addition to PCDH9 and PCDH19, constitutive or regulated proteolytic processing has been reported for other PCDHs (Reiss et al 2006; Buchanan et al 2010; Bouillot et al 2011). Notably, the majority of ncPCDHs harbour predicted nuclear localization signals within their cytoplasmic domains (Figure 4). This raises the possibility that activity-dependent cleavage and subsequent nuclear translocation represent a conserved signalling strategy across the ncPCDH family, coupling synaptic activity to gene regulation, with potential relevance to ASD. Importantly, the marked sequence diversity of ncPCDH intracellular domains suggests that individual family members may engage distinct nuclear partners and modulate gene expression in a PCDH-specific manner. Future studies should address the role of ncPCDH in the regulation of activity-dependent gene expression.
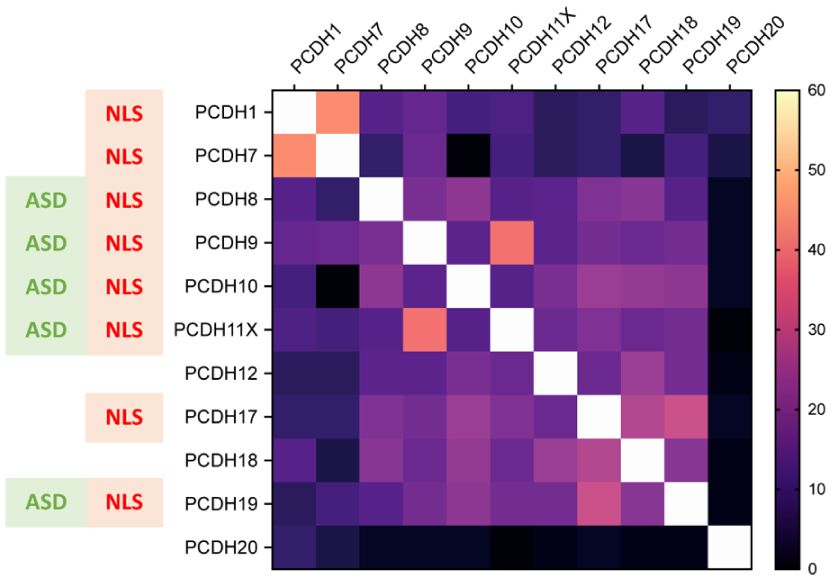
Predicted NLS and sequence similarity across nonclustered PCDH intracellular domains. Pairwise global alignment of human nonclustered PCDH intracellular domain protein sequences was performed using EMBOSS Needle (https://www.ebi.ac.uk/jdispatcher/psa), and percentage identity values are presented in the matrix. The average identity across all comparisons was 14%. NLS motifs were predicted using NLStradamus (http://www.moseslab.csb.utoronto.ca/NLStradamus/), and their presence is indicated for each protein. PCDHs genetically associated to ASD are also marked to highlight the potential relationship between nuclear localization capacity and ASD linkage. ASD, autism spectrum disorder; NLS, nuclear localization signal; PCDH, protocadherin.
Taken together, the activity-driven induction and nuclear translocation of ncPCDHs point to a central role in activity-dependent regulation, a key mechanism implicated in ASD. This signalling network may represent the hub through which ncPCDHs orchestrate synaptic refinement, pruning, circuit assembly, and E/I balance, thereby translating gene-level perturbations into multiscale circuit dysfunction in ASD.
Concluding Remarks
Over the past years, converging evidence from human genetics and mouse models has increasingly implicated ncPCDHs in ASD, linking genetic risk in patients to disrupted neurodevelopmental processes and behaviours in rodents. This progress is most evident for Pcdh9 and Pcdh10, whose mouse mutants serve as informative models recapitulating discrete ASD-relevant domains, alongside Pcdh19, the most extensively studied ncPCDH due to its causal role in DEE9.
Despite these advances, substantial gaps remain. Even in ncPCDH mouse lines that have been characterized in greater depth, behavioural phenotyping has often addressed only a subset of ASD-related traits, while core domains such as social communication and repetitive behaviours remain insufficiently explored. In addition, contrasting outcomes in Pcdh10 and Pcdh19 mutants highlight the need for careful reassessment, and the male bias in ASD prevalence underscores the importance of evaluating both sexes. Finally, ASD-associated Pcdh8 and Pcdh11x remain poorly investigated in vivo, emphasizing the need for dedicated rodent models and systematic behavioural analyses.
At the molecular level, ncPCDH interactors remain largely unknown. Proteomic studies could map these interactions, place them within pathways altered in ASD, and identify potential therapeutic targets. Beyond neurons, PCDHs are expressed in glial populations, yet their functions in these cells remain unexplored. Given the increasing evidence for microglial and astrocyte involvement in ASD, particularly through roles in synaptic pruning and neuroinflammatory signalling (Faust et al 2021), elucidating PCDH functions in glia represents an important direction for future research.
No pharmacologic treatment currently targets core ASD symptoms, with existing therapies limited to behavioural intervention, highlighting the need for drug development (Hirota and King 2023). Rodent models of syndromic ASD have been instrumental for testing therapeutic strategies that subsequently advanced to clinical trials, such as mGluR5 inhibitors, GABAB agonists, and mTOR inhibitors (Jiang et al 2022). In this context, ncPCDH mutant mice may represent valuable additions, particularly in light of the marked genetic and phenotypic heterogeneity of ASD. On one hand, individual ncPCDHs often display region- or circuit-specific expression, making them well suited for dissecting mechanisms underlying defined behaviours and informing targeted therapies for patients with mutations in the corresponding gene. Moreover, as ncPCDHs play key roles during early neurodevelopment, these models may help identify critical developmental windows for intervention. On the other, combining mutations across different ncPCDHs that individually affect distinct ASD-relevant domains—such as sensorimotor dysfunction in Pcdh9 mutants; social communication deficits in Pcdh10 models; and repetitive behaviours, cognitive impairment, and seizure vulnerability in Pcdh19 lines—could yield integrative models that better capture the multifaceted nature of ASD. Such models would also allow the evaluation of therapeutic strategies with broader translational relevance.
Supplemental Material
sj-docx-1-nro-10.1177_10738584261457052 – Supplemental material for Nonclustered Protocadherins in Autism: Integrating Cell Adhesion and Activity-Dependent Signalling
Supplemental material, sj-docx-1-nro-10.1177_10738584261457052 for Nonclustered Protocadherins in Autism: Integrating Cell Adhesion and Activity-Dependent Signalling by Federico Miozzo in The Neuroscientist
Footnotes
Author Contributions
F.M. wrote all the sections and prepared the figures. The author used ChatGPT for English grammar revision and figure refinement.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.