
Abstract
Synaptic dysfunction is a central feature of many neurologic diseases, and the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex plays a critical role in regulating synaptic vesicle fusion and neurotransmitter release. Despite the rapidly growing body of research on SNAREs, comprehensive reviews addressing their mechanisms, as well as the influence of genetic variants and epigenetic regulation, remain limited. This review aims to address this gap. Disruption of SNARE processes can impair synaptic signaling and lead to neurologic pathology. Genetic variants in SNARE-related genes, including VAMP2, STX1A/STX1B, SNAP-25, STXBP1, UNC13A, SYT1, RIM, and RAB3, have been associated with a broad spectrum of neurologic conditions. In addition to genetic variants, emerging evidence indicates that epigenetic mechanisms can regulate the function of SNARE-related genes in physiologic processes and contribute to disease pathogenesis. Genetic variants are increasingly used as diagnostic markers and may inform the development of targeted therapeutic strategies, whereas epigenetic signatures hold promise as diagnostic, prognostic, and treatment-monitoring biomarkers. Although clinical applications remain limited, advancing knowledge of SNARE genetics and epigenetics may facilitate the development of novel diagnostic modalities, prognostic tools, and precision therapeutic strategies for neurologic diseases.
Introduction
The global burden of neurologic disorders increased significantly between 1990 and 2021, rising from approximately 540.25 million cases to 823.80 million cases. The increase is mainly attributed to demographic changes, with population growth accounting for 64.21% and population aging for 35.33%. Among these conditions, headache disorders have the highest incidence, followed by dementia, seizures, and movement disorders, along with various other neurologic diseases (Han et al 2025).
During normal aging, synaptic loss occurs gradually, whereas in pathologic conditions involving cognitive decline, synaptic loss is substantially accelerated and associated with more severe cognitive impairment (Wang et al 2025). Most intracellular membrane fusion events are mediated by soluble N-ethylmaleimide–sensitive factor attachment protein receptors (SNAREs), which also play a central role in neurotransmitter release. During membrane fusion, vesicle-associated SNAREs interact with target membrane SNAREs. Together, these proteins assemble into a SNARE complex that forms a stable 4-helix bundle, providing the mechanical force required to drive membrane fusion.
To achieve physiologic conditions, SNARE function is influenced by genetic and epigenetic mechanisms. Genetic factors provide the template for gene expression, while epigenetic mechanisms regulate whether genes are transcriptionally active or repressed. Epigenetic regulation plays an important role; for example, histone acetylation facilitates the binding of the transcription factor Sp1 to the STX1 promoter, thereby promoting transcription. In contrast, histone deacetylases repress transcription (Nakayama et al 2016).
Although SNAREs play essential roles in normal physiology, their dysfunction may result from genetic variants or other mechanisms, including epigenetic abnormalities, which can contribute to neurologic disorders leading to SNAREopathy (Nakayama et al 2024). SNAREopathy can be considered a specific subgroup within the broader category of synaptopathies. Synaptopathies encompass all disorders caused by defects in synaptic structure or function, whereas SNAREopathy is characterized by dysfunction of the SNARE complex and its regulatory proteins, which directly impair synaptic vesicle docking, priming, and membrane fusion (Verhage and Sørensen 2020). Clinically, SNAREopathy is associated with a wide spectrum of manifestations: early-onset neurodevelopmental disorders, visual impairment, movement disorders, pain syndromes, seizures, developmental delay, speech impairment, psychiatric disorders, and intellectual disability (Guzikowski et al 2025).
In this review, we discuss the mechanisms of SNARE-mediated neurotransmitter release, the disease conditions associated with its dysfunction, genetic variants affecting SNARE, and the epigenetic regulation of SNARE in physiologic and pathologic conditions related to neurologic disorders.
Mechanism of SNARE in Neurotransmitter Release
Membrane fusion is a looping process through 4 essential steps: disassembly, priming, fusion, and recycling (Figure 1). During the disassembly phase, the Munc18-1–syntaxin-1A complex adopts a closed configuration that prevents premature SNARE complex assembly. Munc18-1 (also known as STXBP1) sequesters syntaxin-1A in a “closed” configuration. Munc13-1 catalyzes the transition of the syntaxin-1A into an open, fusion-competent state, thereby enabling Syb2/VAMP2 (vesicle-associated membrane protein 2, also known as synaptobrevin-2) to bind to the open syntaxin-1A complex (Li et al 2018). Together, Munc13-1 and Munc18-1 cooperate to promote proper SNARE complex assembly.
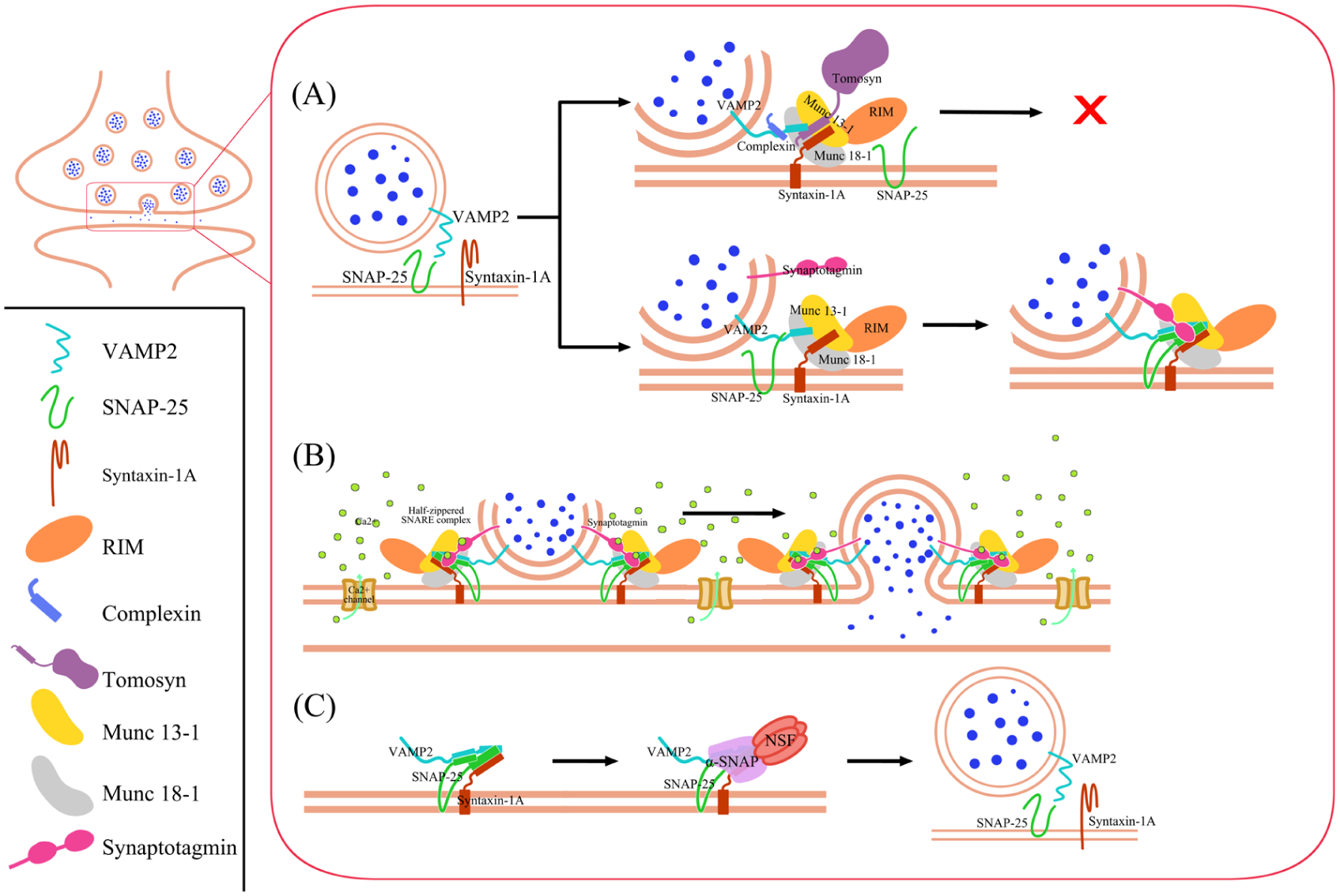
Mechanism of SNARE in neurotransmitter release. The mechanism of SNARE-mediated neurotransmitter release involves 4 main stages: disassembly, priming, fusion, and recycling. The SNARE complex plays a central role throughout these stages. (A) During disassembly and priming, regulatory proteins such as tomosyn and Cpx prevent premature fusion, thereby limiting spontaneous neurotransmitter release. Munc18-1 stabilizes syntaxin-1 A in a closed configuration, while Munc13-1 promotes its transition to an open state and facilitates proper SNARE complex assembly with VAMP2 and SNAP-25. RIM1/2 proteins help localize Munc13-1 at the presynaptic active zone, where synaptic vesicles become primed and contain partially zippered SNARE complexes stabilized by synaptotagmin. (B) During fusion, Ca2+ binding induces configurational changes in Munc13 that position it at the plasma membrane, while synaptotagmin-1 acts as a Ca2+ sensor to trigger full SNARE zippering and synchronous neurotransmitter release. (C) In the recycling stage, α-SNAP binds to SNARE complexes and promotes their disassembly by NSF, enabling the components to be reused and efficiently reassembled into new trans-SNARE complexes with VAMP2 in coordination with Munc18-1, Munc13-1, Cpx, and synaptotagmin. NSF, N-ethylmaleimide–sensitive factor; SNARE, soluble N-ethylmaleimide–sensitive factor attachment protein receptor.
The MUN domain of Munc13-1 facilitates the correct parallel alignment of the syntaxin-1 complex and Syb2/VAMP2 during formation of the trichotomic SNARE complex (Lai et al 2017). In addition, the MUN domain of Munc13-1 binds synaptosomal-associated protein 25 kDa (SNAP-25; Kalyana Sundaram et al 2021). This activity prevents the formation of improper subconfigurations, such as antiparallel arrangements, which represent kinetically trapped dead-end states that do not support efficient synaptic vesicle fusion. Moreover, SNAP-25 together with Munc18-1 and Munc13-1 ensures the correct syntaxin-1A/SNAP-25 subconfiguration within the assembling SNARE complex (Lai et al 2017).
To reduce spontaneous neurotransmitter release and accelerated synaptic depression, tomosyns regulate membrane fusion by increasing the energetic barrier to vesicle fusion, thereby reducing the frequency of fusion events. Mechanistically, tomosyns bind to Syb2/VAMP2-containing template complexes, preventing SNAP-25 association and subsequent SNARE complex assembly (Meijer et al 2024). Not only does syntaxin-1A undergo transitions between closed and open configurations, but Munc13-1 also dynamically switches between distinct configuration states. However, these configurational changes serve different functional roles in the 2 proteins (Grushin et al 2022). Similar to tomosyn, complexin (Cpx) prevents premature SNARE complex zippering and restricts spontaneous exocytosis through its interaction with Syb2/VAMP2 (Vasin et al 2016).
Rab3-interacting molecule 1 and 2 (RIM1/2) proteins play a critical role in the localization and organization of Munc13-1 at the presynaptic active zone. In addition, RIM1/2 recruits synaptic vesicles through its interaction with RAB3 (Persoon et al 2019). During the priming phase, phosphatidylinositol 4,5-bisphosphate (PIP₂) is converted into diacylglycerol (DAG). DAG then binds to Munc13-1, inducing a conformational change that reorganizes the protein into a more compact configuration. This structural rearrangement facilitates synaptic vesicle priming by promoting the assembly of partially zippered SNARE complexes. At this stage, the SNAREs are maintained in a clamped state by synaptotagmin-1 (Syt1) until calcium-triggered neurotransmitter release occurs (Grushin et al 2022).
Calcium (Ca2+) binds to the Munc13 C2B domains, triggering rotation toward the plasma membrane and insertion of Ca2+-binding loops into the lipid bilayer. This rearrangement rotates the MUN-C2C region into direct contact with the plasma membrane. When Ca2+ is later pumped out, half-zippered SNARE complexes remain clamped by Syt1 to form Syt1-SNARE complexes (Grushin et al 2022). During the fusion phase, Syt1 functions as the Ca2+ sensor that triggers synchronous neurotransmitter release. Before Ca2+ influx, the Syt1 C2B domain is bound to the SNARE complex, magnesium (Mg2+), and the plasma membrane, while Cpx binds on the opposite side to clamp membrane fusion (Grushin et al 2019; Voleti et al 2020). Upon Ca2+ in the presynapse, Ca2+ binding to the C2 domains likely displaces the Mg2+ (Grushin et al 2019).
Ca2+- and PIP₂-dependent binding of both Syt1 C2 domains to the plasma membrane releases their interaction with SNARE and bridges the vesicle and plasma membranes. This helps complete SNARE zippering at the C-terminal end and, along with membrane bending caused by the C2 domain, drives membrane fusion and neurotransmitter release (Voleti et al 2020). Neurotransmitter release is tightly regulated by feedback mechanisms involving pre- and postsynaptic components. When postsynaptic signaling is reduced, compensatory retrograde signals can enhance presynaptic neurotransmitter release to restore synaptic strength. Conversely, when neurotransmitter release or postsynaptic activity is excessive, negative feedback mechanisms act to suppress further release and maintain synaptic homeostasis. These bidirectional feedback processes are essential for preserving stable and efficient communication between neurons (Sun et al 2024).
During the recycling stage following neurotransmitter release, α-SNAP acts as an adaptor protein that binds to SNARE complexes and facilitates their recognition and disassembly by NSF (N-ethylmaleimide–sensitive factor). This recycling process enables efficient formation of trans-SNARE complexes with Syb2/VAMP2 on incoming synaptic vesicles in coordination with Munc18, Munc13, Cpx, and Syt1 (White et al 2025). A single SNARE complex remains sufficient to drive fusion with other liposomes or with purified synaptic vesicles. Moreover, multiple SNARE complexes do not act cooperatively during membrane fusion (Van Den Bogaart et al 2010).
SNAREopathy-Associated Variants in Neurologic Disease
Research on the effects of genetic variants in SNAREopathy is ongoing, but several aspects remain underexplored. Variants in SNARE-related genes have been associated with a broad spectrum of neurologic disorders in humans: early-onset neurodevelopmental disorders, visual impairment, movement disorders, pain syndromes, seizures, developmental delay, speech impairment, psychiatric disorders, and intellectual disability. However, despite these clinical associations, the underlying pathogenic mechanisms have been investigated primarily in preclinical studies (Table 1). The common underlying mechanism in these disorders is impaired presynaptic neurotransmission at nerve terminals. This dysfunction leads to a broad spectrum of often overlapping clinical features (Salpietro et al 2019).
Disease-Associated Variants in SNARE-Related Genes Reported in Human Studies.
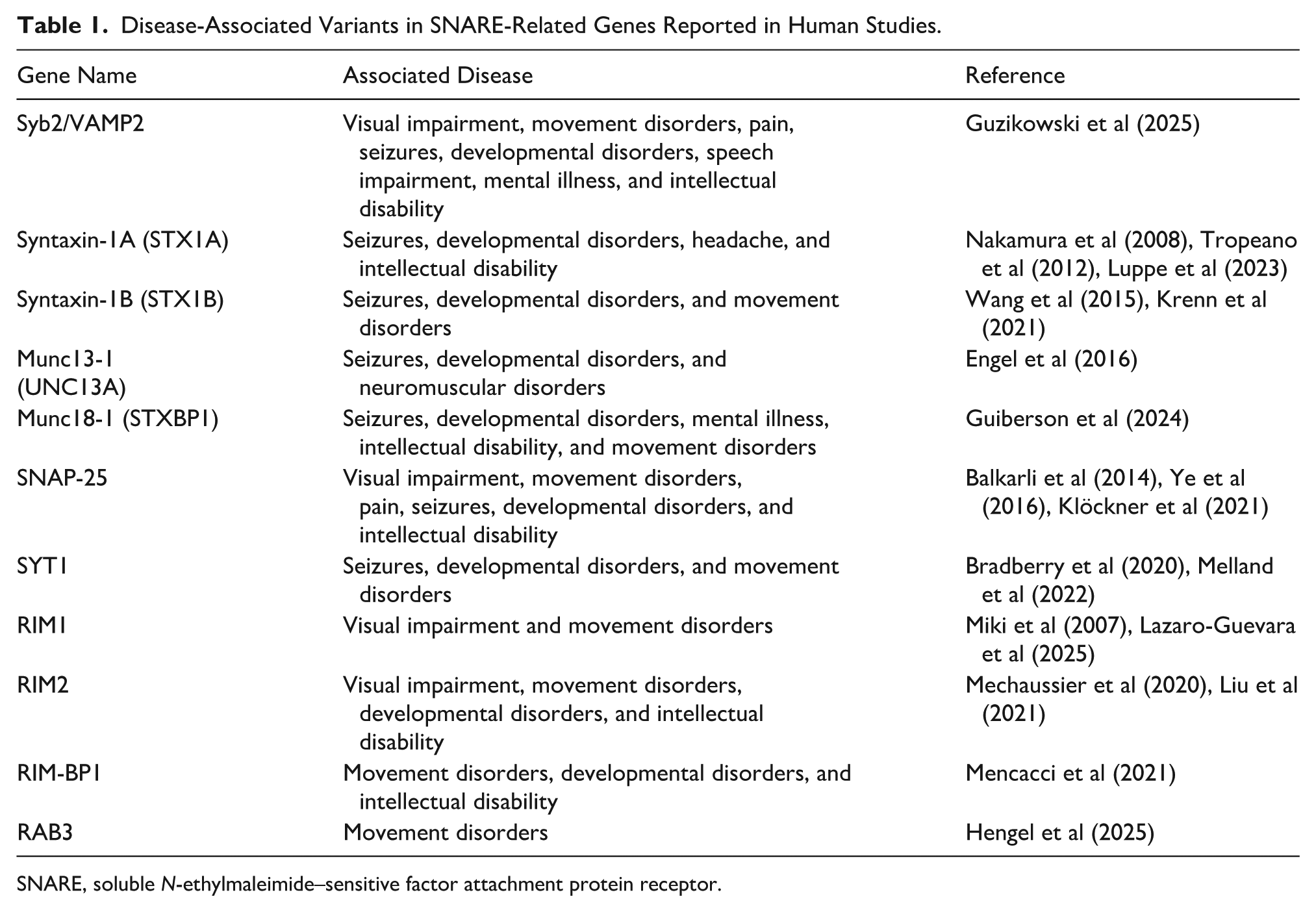
SNARE, soluble N-ethylmaleimide–sensitive factor attachment protein receptor.
As is widely recognized, these variants can alter the regulation and function of SNARE-associated proteins, leading to conditions collectively referred to as SNAREopathy. In neurologic diseases, several SNARE-related variants that remain under investigation have been reported to exhibit structural alterations, aberrant interactions with partner proteins, alterations in splicing regions, and impaired protein stability (Figure 2). These alterations can disrupt synaptic vesicle fusion mechanisms, leading to either increased or decreased neurotransmitter release. Although some variants may influence gene expression, many primarily affect protein structure and function (Mechaussier et al 2020; Vardar et al 2020; Park et al 2024).
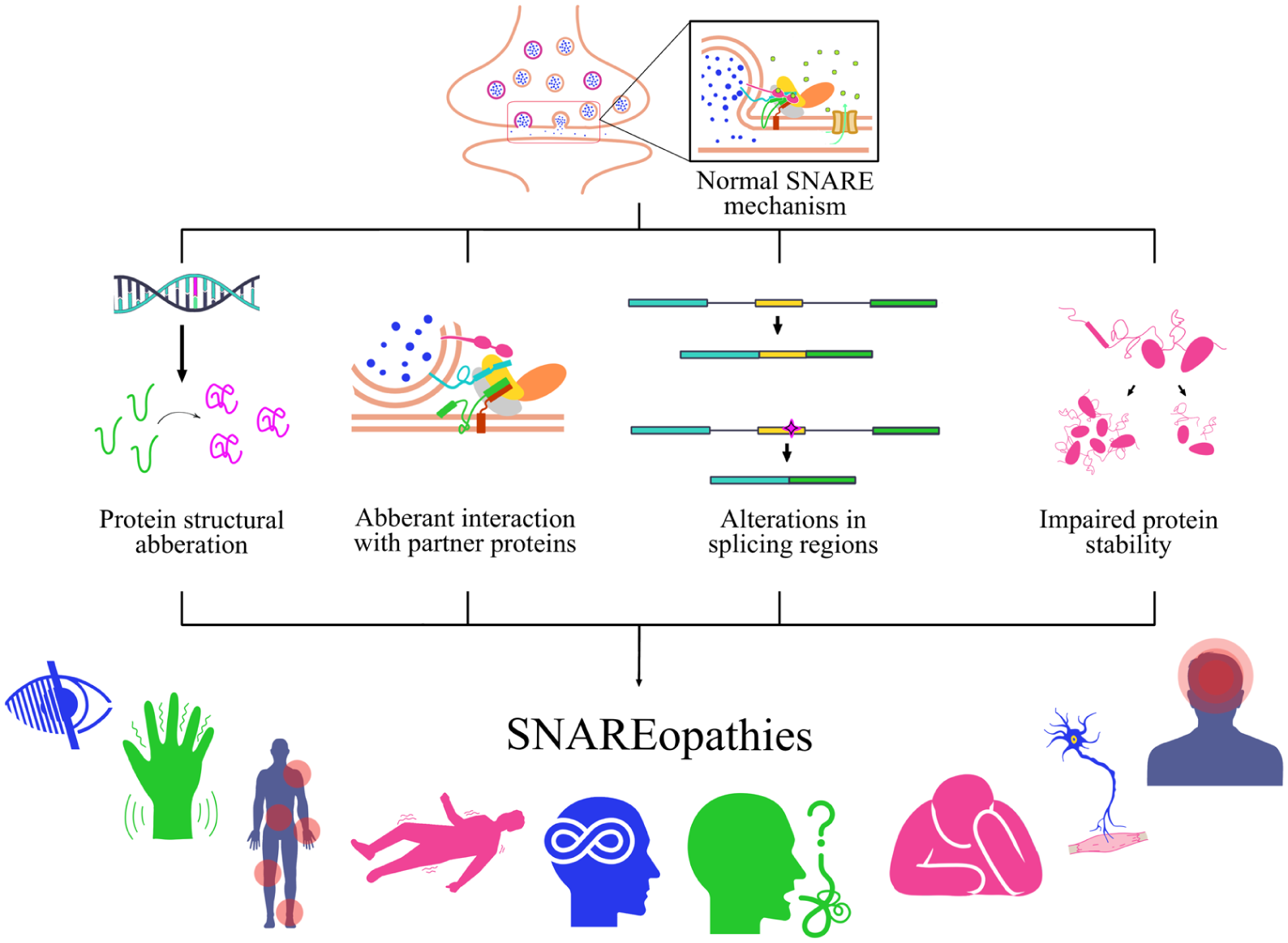
Effects of SNARE-related genetic variants in neurologic disorders. Research on the impact of genetic variants in SNAREopathy is still evolving, with several areas remaining insufficiently understood. In neurologic disorders, a number of SNARE-related variants under investigation have been reported to exhibit protein structural changes, aberrant interactions with partner proteins, alterations in splicing regions, and impaired protein stability. These alterations can impair synaptic vesicle fusion, resulting in increased or decreased neurotransmitter release. As a result, such variants may produce loss- or gain-of-function effects, ultimately contributing to a group of neurologic disorders collectively known as SNAREopathy, which arise from mutations that disrupt the SNARE complex function. SNARE, soluble N-ethylmaleimide–sensitive factor attachment protein receptor.
Current evidence has primarily focused on how the variants affect SNARE complex formation, neurotransmitter release, and related functional outcomes, including spontaneous release. However, their role in disease pathogenesis, particularly how the variants influence brain structure and phenotype, has not yet been thoroughly investigated or discussed.
VAMP2
Variants in VAMP2 have differential effects on SNARE function. These variants can manifest with a range of clinical phenotypes, including visual impairments, movement disorders, pain, seizures, developmental delays, speech impairments, mental illnesses, and intellectual disabilities. Reported variants include p. Val43del (V43del), p. Ile45del (I45del), p. Arg56Leu (R56L), p. Ala67Pro (A67P), p. Gly73Trp (G73W), p. Ser75Pro (S75P), p. Phe77Ser (F77S), and p. Glu78Ala (E78A) (Salpietro et al 2019; Guzikowski et al 2025)
In human studies, mutations in the Syb2/VAMP2 protein were associated with delayed myelin maturation and reduced posterior cerebral white matter volume. In addition, hypoplasia of the optic nerves and optic chiasm has been reported (Salpietro et al 2019), although not all mutations lead to these abnormalities, as some variants are associated with normal anatomy. However, the underlying mechanisms by which these mutations result in structural changes remain poorly understood. The genetic variants V43del, I45del, R56L, A67P, and F77S all exhibited reduced binding affinity to key SNARE-associated proteins, including syntaxin-1A, SNAP-25, and px-1/2. These structural changes in Syb2/VAMP2, a key part of the vesicle fusion machinery, strongly affect synaptic neurotransmitter release and neuronal signaling (Salpietro et al 2019; Guzikowski et al 2025).
Electrophysiologic analyses demonstrated that the N-terminal variants V43del, I45del, and R56X caused a substantial decrease in the frequency of excitatory postsynaptic currents (EPSCs), indicating impaired spontaneous glutamatergic vesicle release. In contrast, the R56L variant did not lower EPSC frequency, although it did change the likelihood of evoked neurotransmitter release. Several C-terminal variants (A67P, S75P, and F77S) that reduced evoked release probability also led to a clear drop in EPSC frequency. By comparison, the G73W and E78A variants caused a strong increase in EPSC frequency, consistent with higher levels of spontaneous synaptic vesicle fusion. This dysregulated spontaneous neurotransmission may be sufficient to contribute to disease pathogenesis, as demonstrated by the Syb2 variants G73W and E78A (Salpietro et al 2019; Guzikowski et al 2025).
STX1
Variants in STX1A have not been extensively investigated, despite evidence that certain variants are associated with disease. One study, although not examining clinically identified variants, reported that the STX1A L165A/E166A substitutions did not alter the global configuration of the Munc18a–syntaxin- 1A complex but resulted in a modest reduction in Munc18a binding affinity. This reduction is thought to arise because variants lacking the N-peptide or carrying the LE mutation bind to Munc18a in a closed configuration. The N-peptide of syntaxin 1A (amino acids 1–24) has been implicated in facilitating the transition from the Munc18a-bound syntaxin 1A state to the Munc18a-bound SNARE complex, even though these alterations do not disrupt the overall configuration. However, this study did not further investigate the impact on exocytotic function (Colbert et al 2013).
Additional STX1A variants, including Cys145Trp, Val223del, Val241del, and Met79Arg, are associated with neurodevelopmental disorders with or without epilepsy but remain poorly characterized. To date, available evidence is largely limited to bioinformatic analyses, which suggest that these variants may alter protein structure and potentially affect interactions with Munc18-1 (Luppe et al 2023). In contrast, several variants in STX1B (c.133_134insGGATGTGCATTG; p.Lys45delinsArgMetCysIleGlu; c.135_136AC>GA; p.Leu46Met; c.166C>T; p.Gln56*) have been associated with normal GABAergic and glutamatergic motor cortical excitability, despite a history of febrile seizures and epilepsy that has since remitted (Stefanou et al 2017).
Another study investigated the variants p.Lys45delinsArgMetCysIleGlu, p.Gly226Arg, and p.Val216Glu. The p.Lys45delinsArgMetCysIleGlu variant destabilized the protein, promoted STX1B degradation, and severely impaired its expression and trafficking to synapses, leading to an inability to sustain neurotransmitter release. It was also associated with reduced neuronal survival, similar to some epileptogenic Munc18-1 mutations, as well as increased protein misfolding and enhanced spontaneous neurotransmitter release (Vardar et al 2020). In contrast, the p.Gly226Arg and p.Val216Glu variants appeared relatively stable but exhibited altered interactions with Munc18-1. Specifically, p.Gly226Arg resulted in an approximate 50% reduction in the expression levels of STX1B and Munc18-1 proteins and decreased the readily releasable pool of synaptic vesicles. Meanwhile, p.Val216Glu only mildly reduced interactions with Munc18-1 and Munc13 but increased vesicle fusogenicity and release probability (Vardar et al 2020).
Munc
The p.Pro814Leu variant in Munc13 has been shown to increase synaptic vesicle fusogenicity. This effect is mediated by enhanced Ca2+ affinity of the C2B domain, which promotes an elevated probability of synaptic vesicle fusion. Munc13-1 comprises 3 regulatory domains: a Ca2+–calmodulin (CaM) binding domain, a C1 domain, and a central C2 domain (C2B; Lipstein et al 2017). Similarly, the Munc18 variant p.Leu446Phe is associated with an approximate twofold increase in the number of synaptic vesicles fusing with the plasma membrane in response to a single action potential as compared with control neurons, despite only minor changes in neuronal morphology and synapse density (Lammertse et al 2020).
Additional Munc18 variants exhibit diverse functional effects. The M443R and G544D variants are associated with reduced synaptic density, whereas the R388X and V84D variants fail to rescue neuronal viability and show mild impairments in synapse formation or maintenance. In contrast, the C180Y and C522R variants display relatively normal synaptic transmission. Notably, C180Y, T574P, and C522R demonstrate reduced protein stability, while M443R and G544D impair the synaptic targeting of Munc18-1 and syntaxin-1A. These defects may contribute to the observed reduction in synapse density (Kovačević et al 2018). Destabilization of Munc18-1 promotes secondary co-aggregation of its interacting partners, including syntaxin-1A, Doc2A, and Doc2B. Furthermore, it has been demonstrated that Munc18-1 missense variants induce aggregation of Doc2A and Doc2B and that loss of functional Munc18-1 compromises the synaptic trafficking of Doc2A/B, indicating a critical role for Munc18-1 in maintaining the stability and proper localization of these presynaptic proteins (Guiberson et al 2024).
SNAP-25
Variant Ser187Ala in SNAP-25 induces an immature phenotype in dentate granule cells in adult mice. This alteration leads to an overall enlargement of the hippocampal dentate gyrus, including expansion of the hilar region. Granule cells exhibit a reduced threshold current for action potential generation and produce a greater number of action potentials during sustained depolarization, indicating enhanced intrinsic excitability (Ohira et al 2013). Furthermore, this substitution affects working memory. These changes are associated with working memory deficits resembling those observed in neuropsychiatric disorders such as schizophrenia, attention-deficit/hyperactivity disorder, and anxiety disorders and are accompanied by increased susceptibility to epilepsy (Ohira et al 2013).
Other SNAP-25 variants, including V48F and D166Y, disrupt the Syt1-binding interface, whereas I67N destabilizes the SNARE complex. All 3 variants impair Syt1-dependent vesicle docking and Ca2+-triggered fusion. V48F and D166Y, with D166Y exhibiting a stronger effect, demonstrate by increased spontaneous release, reduced readily releasable pool size, elevated release probability, and enhanced spontaneous SNARE assembly, ultimately leading to dysregulated membrane fusion (Kádková et al 2024). In contrast, I67N reduces spontaneous and evoked neurotransmitter release by increasing the energy barrier for fusion, without affecting the readily releasable pool size (Kádková et al 2024).
SYT1
SYT1 variants alter the structure of the Ca2+-binding pocket. Specifically, the D304G variant removes an acidic Ca2+-coordinating residue; D366E preserves the acidic ligand but reduces the size of the binding pocket; and I368T decreases the hydrophobicity of the membrane-penetrating tip within a Ca2+-binding loop. Functionally, D303G and I367T cause stronger impairments in evoked synaptic release than D365E, indicating a graded dominant-negative effect (D303G ≈ I367T > D365E) that aligns with the severity of clinical symptoms. All variants exhibit reduced binding to PIP2-containing membranes under Ca2+-dependent conditions. In addition, Ca2+ dissociates more rapidly from mutant C2AB domains, while mutant C2A domains display decreased Ca2+ affinity, consistent with impaired membrane engagement and synaptic vesicle exocytosis (Bradberry et al 2020).
A distinct missense variant, P401L in the C2B domain, was shown to reduce dendritic outgrowth, accompanied by a proportional decrease in synapse number. Action potentials caused over a twofold increase in asynchronous release, resulting in less coordinated neurotransmission. This appears to stem from a larger readily releasable pool for asynchronous release and weaker suppression of spontaneous and asynchronous fusion. Neurons appear to compensate for this heightened synaptic input by shortening their dendrites. This variant acts in a dominant-negative manner, supporting its pathogenic role in the heterozygous patient. Mechanistically, substitution of a rigid proline with a more flexible leucine at the base of the C2B domain is proposed to impair clamping of vesicle release, disrupting the main interaction between Syt1 and the SNARE complex (van Boven et al 2024).
Most variants within the C2A (L159R, T196K, E209K, E219Q) and C2B (M303V, S309P, Y365C, G369D) domains of SYT1 do not alter overall protein expression or neuronal localization. However, the L159R variant markedly reduces synaptic SYT1 levels. Variants M303V and S309P in the C2B domain significantly decrease the size of the synaptic vesicle recycling pool, whereas C2A variants and the remaining C2B variants do not affect total vesicle availability for exocytosis. Variants in the C2A and C2B domains impair evoked exocytosis of recycling pool vesicles through a dominant-negative effect. These variants lead to varying degrees of reduced neurotransmitter release, reflected in how often they differ significantly from the wild type; notably, T196K shows no significant difference from the wild type. Overall, C2B domain variants exert more pronounced effects than C2A variants. All C2B variants significantly slow the initial rate of exocytosis, with varying degrees of impairments: M303V and S309P reduce the initial exocytic rate to a degree comparable to the I368T reference variant, whereas Y365C and G369D show milder effects. C2A variants produce a modest nonsignificant reduction in initial exocytic rate. Furthermore, C2B variants exert varying degrees of effects on the readily releasable pool, with significant reductions observed for M303V, S309P, and I368T and a trend toward reduction for Y365C. In contrast, C2A variants do not affect readily releasable pool size (Park et al 2024).
RIM
RIM proteins have also been implicated in visual system disorders. Current evidence demonstrates that deletion of RIM1/2 is sufficient to reduce presynaptic release probability onto neurons of the dorsal lateral geniculate nucleus (dLGN). In RIM1/2 conditional knockout mice, eye-specific segregation within the dLGN and topographic refinement of ipsilateral retinal ganglion cell axons in the dLGN and superior colliculus (SC) are impaired, recapitulating abnormalities observed following global disruption of retinal activity. In contrast, reduced synaptic release does not alter eye-specific lamination in the SC or the retinotopic refinement of contralateral retinal ganglion cell axons in the SC (Assali et al 2017).
The RIM1 R655H variant heightens RIM1-dependent modulation by significantly increasing the noninactivating component of Cav2.1 channel currents, enhancing current density, accelerating channel activation, and further suppressing inactivation. Collectively, these effects are expected to elevate Ca2+ influx and consequently promote increased neurotransmitter release and synaptic transmission. In contrast, for L-type Cav1.4 channels, which serve as the primary mediators of glutamate release at photoreceptor ribbon synapses, the mutation abolishes the RIM1-mediated hyperpolarizing shift in voltage-dependent activation. Notably, activation speed and current density are similar to those seen in wild type RIM1. However, the loss of this modulatory effect is likely to impair synaptic transmission in the visual system (Miki et al 2007).
In the RIM2 variant c.4363+1G>A, bioinformatic analyses predict skipping of the adjacent exon, resulting in a frameshift and the introduction of a premature termination codon. This alteration is therefore expected to produce a truncated protein or trigger nonsense-mediated mRNA decay (Mechaussier et al 2020). However, when compared with RIM1 variants, this mutation has not been extensively functionally characterized. The RIMBP1 p.Gly1808Ser variant is a pathogenic missense mutation associated with altered synaptic function. This variant produces approximately threefold-larger spike-evoked calcium transients due to increased Cav2.1 channel recruitment at presynaptic release sites, leading to enhanced neurotransmission through elevated Ca2+ influx (Mencacci et al 2021).
RAB3
The Rab3 variants p.Arg82Trp and p.Tyr90Cys disrupt normal synaptic structure and function. These variants cause enlargement of the presynaptic active zone precursor Bruchpilot and increase the number of postsynaptic glutamate receptor clusters that are not properly aligned with Bruchpilot. Both mutations reduce RAB3GAP1-mediated GTP hydrolysis, indicating impaired Rab3 regulation. Additionally, the p.Arg82Trp variant specifically shows reduced interaction with Rab-3A (Hengel et al 2025).
Disease Mechanisms in Epigenetic Regulation of SNARE
Genetic variants have been implicated in SNARE-related pathology, similar to findings in other neurologic disorders. However, epigenetic mechanisms remain comparatively understudied. Epigenetic regulation plays an essential role in normal physiology. For example, histone acetylation is required for the binding of the transcription factor Sp1 to the STX1 promoter, which promotes transcription. In contrast, histone deacetylases repress transcription (Nakayama et al 2016). In addition, transcription factors can function as repressors. For example, the Yin-Yang 1 transcription factor binds to the −183 to −137 region of the STX1 promoter and interacts with histone deacetylases such as HDAC to suppress transcription (Nakayama et al 2021).
Aberrant epigenetic regulation can contribute to pathologic processes, as discussed in this section (Figure 3). Epigenetic states can also be influenced by genetic variants, although this relationship has not been extensively explored in the context of SNARE. Epigenetic regulation broadly includes DNA methylation, RNA methylation, histone modifications, noncoding RNAs, and chromatin remodeling. Among these 5 mechanisms, chromatin remodeling is the only one that has not yet been reported in SNARE-associated diseases.
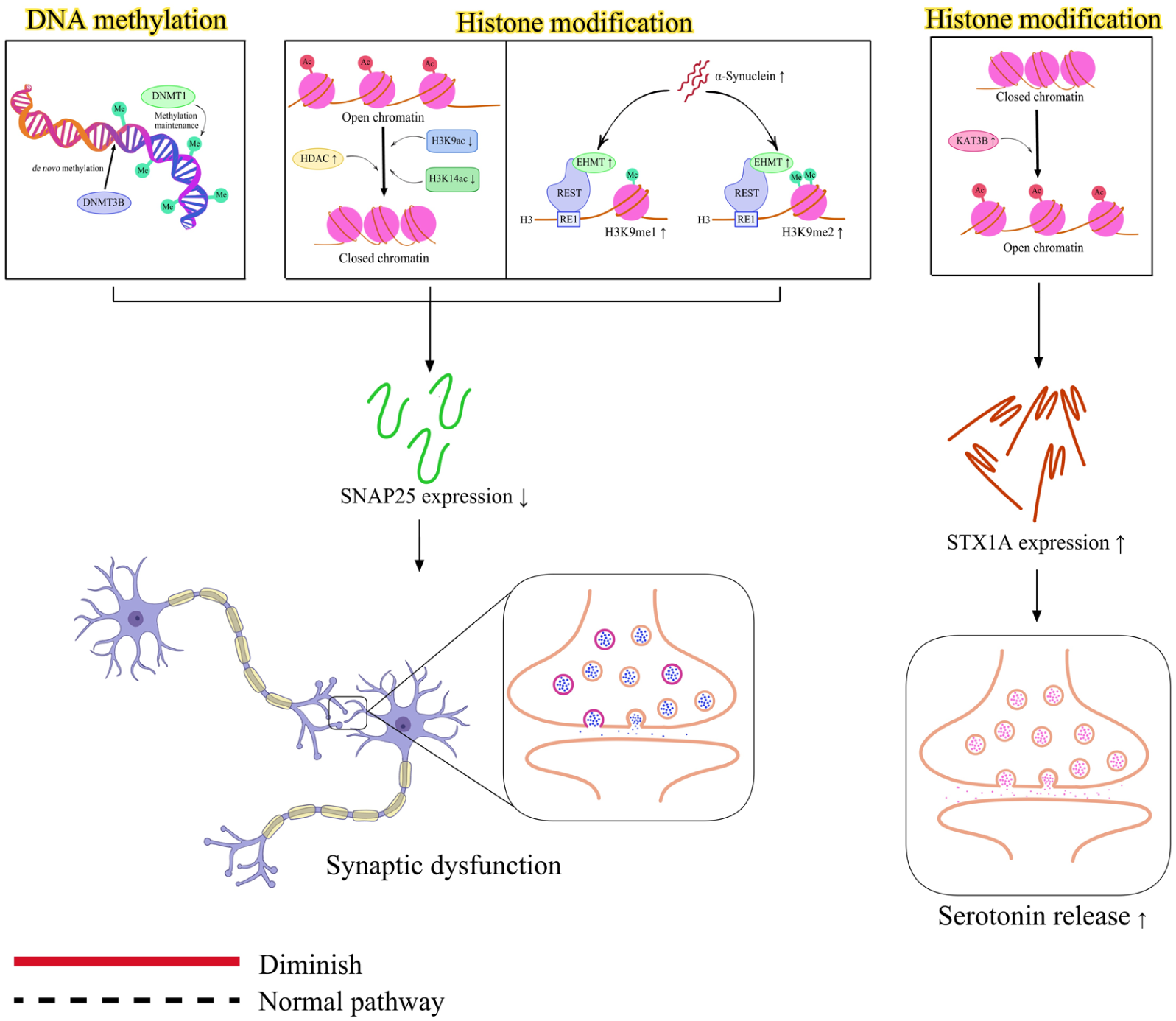
Epigenetic influence in SNAREopathy. Epigenetic regulation of SNARE-related genes involves several mechanisms, including DNA methylation, histone modifications, RNA methylation, and noncoding RNAs. However, chromatin remodeling has not yet been explored in SNARE-related diseases. Increased DNA methylation (via DNMT1 and DNMT3B), as well as histone modifications characterized by increased HDAC2 expression and decreased levels of H3K9ac and H3K14ac, causes decreased SNAP-25 expression. Another histone modification process plays a role in regulating SNAP-25 expression, where α-synuclein upregulates EHMT2, which, in cooperation with the REST transcription factor, represses SNAP-25 transcription through H3K9 methylation, thereby contributing to its reduced expression. The decreased SNAP-25 expression, along with ubiquitination and degradation of the GluN2B, leads to synaptic dysfunction. In contrast, the histone modifications through activation of KAT3B (p300) increase STX1A expression and enhance serotonin release. SNARE, soluble N-ethylmaleimide–sensitive factor attachment protein receptor.
In the case of DNA methylation of SNAP-25, no significant differences have been reported in Alzheimer disease, despite reduced SNAP-25 gene expression in 3 brain regions of patients with Alzheimer disease. However, the DNA methylation analysis in that study examined only 2 regions located at 420 to 445 bp and 792 to 816 bp (Furuya et al 2012). It is therefore possible that methylation changes occur at other CpG sites that were not analyzed, as many CpG sites are not directly associated with specific phenotypes. Alternatively, the reduced SNAP-25 expression may result from other epigenetic mechanisms.
Another study investigating SNAP-25 used a comprehensive approach to examine histone modifications and DNA methylation in the context of neurodegeneration induced by chronic hypoxia. Chronic hypoxia is characterized by glutamate excitotoxicity, activation of apoptotic cascades, reduced synaptic density, and calcium overload in the hippocampus. These changes are linked to higher glutamate levels in synaptosomes, which can lead to excitotoxic damage. The elevated glutamate levels are linked to increased synaptic vesicle density and reduced vesicle fusion. This reduction in vesicle fusion is associated with decreased transcriptional activity due to reduced transcription factor binding to the SNAP-25 promoter. The mechanism appears to involve epigenetic regulation, including increased DNA methylation mediated by DNMT1 and DNMT3B, as well as histone modifications characterized by increased HDAC2 expression and decreased levels of H3K9ac and H3K14ac. These epigenetic alterations contribute to the reduced expression of SNAP-25 observed under chronic hypoxic conditions (Biswal et al 2017).
α-Synuclein is associated with neurodegenerative diseases. Overexpression of α-synuclein increases the expression of euchromatic histone lysine N-methyltransferase 2 (EHMT2). EHMT2 represses gene transcription by increasing H3K9me1 and H3K9me2 levels. With the REST transcription factor, EHMT2 is recruited to the RE1 site in the promoter region of the SNAP-25 gene, where it catalyzes the dimethylation of histone H3 lysine 9 (H3K9me2). This modification results in repression of the SNAP-25 promoter, contributing to synaptic dysfunction in response to α-synuclein exposure (Sugeno et al 2016).
KAT3B, also known as p300 (EP300), is a key lysine acetyltransferase that regulates neuronal STX1A transcription through histone acetylation. In STX1A-deficient mice, activation of KAT3B increases STX1A expression and enhances serotonin (5-HT) release, thereby restoring serotonergic function and improving low locomotor activity behavior (Nakayama et al 2024).
Translational and Future Direction of SNAREopathy
Genetic variants have a more established role in clinical practice than epigenetic alterations, although their application in SNARE-related disorders remains relatively limited. As discussed in Table 1, several variants in SNARE-related genes have been identified in patients and are already useful for diagnosis and, in some cases, for guiding treatment decisions. However, their value as prognostic markers has not yet been clearly established. While the clinical significance of genetic variants has been extensively explored in many disorders, research focusing on SNARE-related variants is still emerging. Nevertheless, available evidence suggests that these variants may have therapeutic implications. For example, the SNAP-25 I67N variant showed a favorable response to the K+-channel blocker 4-aminopyridine in preclinical studies, likely through enhancement of Ca2+ influx (Østergaard et al 2025). A similar approach was later applied in a patient carrying the VAMP2 p.Arg56 variant, where treatment with 4-aminopyridine was associated with improvements in emotional and behavioral regulation as well as cognitive function (Simmons et al 2020).
In contrast to genetic variants, epigenetic alterations are not currently used as diagnostic markers in SNARE-related disorders, despite growing evidence supporting their potential clinical utility. Numerous studies have identified disease-associated epigenetic signatures, including altered microRNA expression (Agostini et al 2023) and altered DNA methylation (Biswal et al 2017), and changes in histone deacetylase–mediated transcriptional regulation of STX1A (Nakayama et al 2021) may be detectable in peripheral blood.
Epigenetic markers may have value in monitoring therapeutic response. In sarcopenia, a condition characterized by alterations at the neuromuscular junction, individuals carrying the SNAP-25 rs363050 AA genotype exhibited significant changes in miRNA expression following a 30-day rehabilitation program. In particular, altered miR-451a expression was observed after rehabilitation, suggesting that this miRNA may serve as a potential biomarker of treatment response. At baseline, miR-451a was significantly upregulated in these patients. Following rehabilitation, miR-451a expression became significantly downregulated in improved disease status. Rehabilitation was further associated with significant upregulation of miR-155-5p, miR-421-3p, miR-425-5p, miR-744-5p, and miR-93-5p, whereas miR-495-3p expression was significantly downregulated (Agostini et al 2021).
However, no data are currently available regarding the direct expression of SNAP-25 rs363050 AA in individuals with sarcopenia. Previous studies have shown that reduced SNAP-25 expression is associated with neurologic abnormalities, including hyperactivity, cognitive impairment, social deficits, and abnormal electroencephalographic patterns (Braida et al 2015). In a separate study, downregulation of miR-451a increased MYC expression through interaction with an miR-451a-binding sequence in the MYC 3′-UTR (Zeng et al 2017).
Although the regulatory relationship between miR-451a and SNAP-25 has not been directly demonstrated, the significant reduction in miR-451a expression following the 30-day rehabilitation program may reflect epigenetic modulation of pathways involved in SNAP-25 regulation and neuromuscular function. Evidence supporting miRNA-mediated regulation of SNAP-25 has been reported in vitro, where miR-181a-5p and miR-23a-3p were shown to bind the SNAP-25 3′-UTR. Interestingly, miR-181a-5p increased SNAP-25 protein expression, whereas miR-23a-3p reduced SNAP-25 protein levels (Agostini et al 2023).
Although epigenetic-based drugs targeting SNARE are not yet widely available, studies in other fields have already demonstrated their potential. For example, T-cell receptor engineering of CD8+ T cells enables the recognition of tumor-associated antigens and tumor-specific neoantigens (Kroonen et al 2024). This approach may provide insight into the development of epigenetic therapies targeting SNARE-related pathways. Beyond therapeutic applications, epigenetic alterations in SNARE may have prognostic value. Epigenetic biomarkers have been successfully used for prognosis in various diseases (Supic et al 2022). However, their prognostic potential SNARE-related pathways remains largely unexplored. Further studies are needed to evaluate whether SNARE-associated epigenetic signatures can be used to predict disease progression and clinical outcomes.
Conclusion
SNAREs are essential for synaptic vesicle fusion and neurotransmitter release, ensuring precise communication between neurons. Genetic variants that disrupt SNARE function can impair synaptic transmission and contribute to a range of neurologic disorders. In addition to genetic factors, growing evidence suggests that epigenetic mechanisms play an important role in regulating SNARE gene expression. Further research is needed to elucidate the molecular mechanisms underlying SNAREopathy and their contribution to neurologic disorders, particularly the epigenetic regulation of SNARE-related pathways.
Footnotes
Author Contributions
V.B.: conceptualization, methodology, formal analysis, investigation, resources, writing–review and editing. L.T.M.: conceptualization, methodology, formal analysis, investigation, visualization, writing–original draft, writing–review and editing. F.M.H.: conceptualization, methodology, formal analysis, investigation, resources, writing–original draft, writing–review and editing.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.