
Abstract
Objective
LncRNA HOXA11-AS was abnormally upregulated during heart failure, suggesting that it might be involved in the development of chronic heart failure (CHF). This study aims to explore the diagnostic value of HOXA11-AS in CHF and its mechanism of action in myocardial injury.
Methods
The level of HOXA11-AS in serum was detected by real-time quantitative polymerase chain reaction (RT-qPCR), and its diagnostic efficacy was evaluated by ROC curve. The model of CHF was established by treating AC16 cells with doxorubicin (DOX). Cell viability and apoptosis were detected by cell counting kit-8 (CCK-8) and flow cytometry. The levels of inflammatory factors and myocardial injury markers were detected by enzyme-linked immunosorbent assay (ELISA). The content of malondialdehyde (MDA) and the activity of superoxide dismutase (SOD) were detected by kits. Dual-luciferase reporter assay was used to verify the targeting relationship between HOXA11-AS and miR-342-3p.
Results
The expression of HOXA11-AS in the serum of CHF patients was significantly increased. ROC analysis showed that HOXA11-AS had a high diagnostic value for CHF. In the DOX-induced cell model, knocking down HOXA11-AS could significantly enhance cell viability, inhibit cell apoptosis, and reduce the release of myocardial injury markers, pro-inflammatory factors, as well as the level of oxidative stress. miR-342-3p was the target gene of HOXA11-AS. Inhibition of miR-342-3p could reverse the myocardial protective effect produced by the knockout of HOXA11-AS.
Conclusion
HOXA11-AS, as a potential biomarker for diagnosing CHF, exacerbates myocardial injury, inflammatory response and oxidative stress by sponging miR-342-3p.
Introduction
Chronic heart failure (CHF) is a clinical syndrome caused by long-term impairment of the heart's pumping function. The core issue lies in the fact that the amount of blood pumped by the heart cannot meet the metabolic demands of the body. 1 CHF not only causes patients to experience breathing difficulties, but also frequently leads to multi-organ dysfunction. Moreover, the mortality rate of this disease is extremely high. 2 Although significant progress has been made in the treatment of heart failure in recent years, 3 its high mortality rate and poor prognosis still indicate that we still need to further explore the targets for its early diagnosis and treatment.
Long non-coding RNAs (LncRNAs) are involved in a range of diseases such as cancer and immune disorders.4–7 In the field of cardiovascular diseases, lncRNAs can affect various processes, such as apoptosis, hypertrophy, fibrosis and metabolic remodeling of cardiomyocytes.8,9 Based on the screening results from the GEO database, the expression level of lncRNA HOXA11-AS increased during heart failure. 10 Atherosclerosis is the core pathological basis of CHF, and HOXA11-AS has also been proven to exacerbate the inflammatory response in diabetes-related atherosclerosis. 11 These findings collectively suggest that HOXA11-AS may play a role in promoting pathological changes during the occurrence and development of CHF, but the specific mechanism still needs to be further elucidated.
MicroRNAs (miRNAs) play a crucial role in the regulation of gene expression and also have significant impacts on cardiovascular diseases.12,13 Among them, miR-342-3p shows abnormal expression in CHF.14,15 miR-342-3p inhibits the death of cardiomyocyte by targeting SOX6 and TFEB. 16 Additionally, this miRNA has been confirmed to have anti-inflammatory effects on cardiovascular diseases. 17 These results imply that miR-342-3p appears to exert cardiac protective functions through multiple pathways, and its downregulation is closely related to the pathological process of CHF.
Based on the above research background, this study aims to explore the diagnostic value of lncRNA HOXA11-AS for CHF, and further investigate the role of the HOXA11-AS/miR-342-3p axis in the occurrence and development of CHF.
Materials and Methods
Participants
This study included a total of 115 patients diagnosed with CHF at Nantong First People's Hospital from 2022 to 2024. At the same time, 110 healthy individuals who underwent health check-ups during the same period and did not have CHF or any other diseases listed as exclusion criteria were selected as the control group. The diagnostic criteria for CHF were applied according to the relevant clinical guidelines.18,19 The inclusion criteria for patients were: (1) diagnosed with CHF; (2) in a stable clinical condition; (3) without major surgical history in the past year. The exclusion criteria for patients and the control group were: (1) suffering from acute coronary syndrome, congenital heart disease, pulmonary heart disease, malignant tumors, etc; (2) having liver or kidney failure; (3) those who have experienced a stroke or acute cerebrovascular disease within the past six months. All participants signed informed consent forms. The study protocol was approved by the Ethics Committee of Nantong First People's Hospital and strictly followed the ethical guidelines of the Helsinki Declaration.
Sample Collection
Fasting venous blood was collected from each subject. The blood samples were immediately subjected to centrifugation (1500 × g, 10 min, 4 °C) to separate the serum. Serum was aliquoted and stored at −80 °C.
Cell Culture and Induction of CHF Model
The human cardiomyocytes (AC16, ATCC, USA) were cultured in DMEM medium (Cat# 12491015, Gibco, USA) supplemented with 10% FBS (Cat# A5256701, Gibco, USA) and 1% penicillin-streptomycin (Cat# P1400, Solarbio, China). These cells were cultured in an incubator at a constant temperature of 37 °C and 5% CO₂. During the cultivation process, a fresh complete culture medium was replaced every 2 to 3 days. When the fusion rate reaches 80%–90%, the cells were digested with 0.25% trypsin-EDTA (Cat# T1300, Solarbio, China) and then passaged at a ratio of 1:4. Cells that had been passaged 3 to 10 times were used for the subsequent research.
To cause damage to the cardiomyocytes, when the cells reach an appropriate density, the culture medium was replaced with a solution containing different concentrations of doxorubicin (DOX, Cat# D1515, Sigma-Aldrich, USA). The treament duration was 24 h. Based on the detection of cell viability, this study ultimately selected 2 μM of DOX as the optimal concentration for inducing cell damage. This concentration could cause a significant decline in cell vitality.
Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated from samples using TRIzol reagent (Cat# 15596018CN, Invitrogen, USA). The RNA was reverse-transcribed into cDNA using the FastKing gDNA Dispelling RT SuperMix (Cat# RR037, TIANGEN, China) and the miRNA cDNA Synthesis Kit (Cat# CW2141, Cwbiotech, China), respectively. Finally, using cDNA as the template, qPCR was carried out with the SuperReal PreMix Plus (Cat# RR430, TIANGEN, China) and miRNA qPCR Assay Kit (Cat# CW2142, Cwbiotech, China). The reaction system consists of 10 μL of pre-mix, 0.6 μL of primers, 2 μL of cDNA template, and 4.8 μL of RNase-free H₂O. The reaction procedure is as follows: 15 min of pre-denaturation at 95 °C; then, conduct 40 cycles (95 °C for 10 s, 60 °C for 30 s). GAPDH and U6 were used as reference genes. The experiment was performed with 5 biological replicates and 3 technical replicates. The sequences of the primers are as follows: miR-342-3p, F: 5′- GGGTCTCACACAGAAATCGC -3′; HOXA11-AS, F: 5′- ACGCTAGGACACCATTTT -3′, R: 5′- CCGGCTACTAGTCAGTGA-3′; GAPDH, F: 5′-GGAGCGAGATCCCCCCCCAAAT-3′, R: 5′- GGCTGTGTCATACTTCATG -3′; U6, F: 5′- GATTATCGGACCACCATTCCACTG -3′; R: 5′- GATCTGGTTCCCAATGACTGTG -3′.
Cell Transfection
The small interfering RNA (si-HOXA11-AS, si-NC) and miRNA inhibitor (miR-342-3p inhibitor, inhibitor NC) were synthesized by GenePharma (China). AC16 cells were seeded at a density of 2 × 105 cells per well. Transfection was performed when the cell confluence reached 60%–70%. During transfection, siRNA or miRNA inhibitors was mixed with Lipofectamine 2000 (Cat# 11668500, Invitrogen, USA) in a volume ratio of 1:2. The mixture was incubated at room temperature for 20 min to form the transfection complex. Then, the complex solution was added to the cells. After 48 h of transfection, the culture medium was replaced with a solution containing 2 μM of DOX to establish a cell model of CHF.
Cell Viability Assay
AC16 cells were seeded in 96-well plates. After the cells adhered to the plates, they were treated with different concentrations of DOX for 24 h. Then, 10 μL of CCK-8 solution (Cat# C0037, Beyotime, China) was added to each well, and the plates were incubated in the incubator for 2 h. Finally, absorbance was recorded at a wavelength of 450 nm using a microplate reader (FLx800, BioTek, USA). The experiment was performed with 5 biological replicates and 3 technical replicates.
Cell Apoptosis Assay
The level of apoptosis was detected using the Annexin V-FITC/PI kit (Cat# 556547, BD Biosciences, USA). After digesting the AC16 cells with trypsin, the cells were washed twice with pre-cooled PBS. Then, the cells were resuspended in 100 μL binding buffer, and were stained with 5 μL of Annexin V-FITC and PI. The cells were incubated at room temperature in the dark for 15 min. Flow cytometry analysis was performed on a flow cytometer (BD FACSCanto II, BD Biosciences, USA). The experiment was performed with 5 biological replicates and 3 technical replicates.
Enzyme-Linked Immunosorbent Assay (ELISA)
The contents of Atrial Natriuretic Peptide (ANP), Brain Natriuretic Peptide (BNP), Interleukin-1β (IL-1β) and Tumor Necrosis Factor-α (TNF-α) were detected by ELISA. Specifically, ANP (Cat# CSB-E11193h) and BNP (Cat# CSB-E07970h) were detected using the kits from CUSABIO (USA), while IL-1β (Cat# E-EL-H0149) and TNF-α (Cat# E-EL-H0109) were measured using the kits from Elabscience (China). The collected cells were lysed. Subsequently, the samples and standard substances were added to the plate coated with antibodies, and incubated at room temperature. And then reacted with biotinylated antibodies and streptavidin. Finally, add TMB and measure the absorbance value at 450 nm. The experiment was performed with 5 biological replicates and 3 technical replicates.
Evaluation of Oxidative Stress
The content of malondialdehyde (MDA, Cat# A001-3-2) in the cells and the activity of total superoxide dismutase (SOD, Cat#A003-1-2) were determined using the corresponding reagent kits from Nanjing Jiancheng Bioengineering Institute (China). AC16 cells were treated with lysis buffer containing 0.2% Triton X-100, and then centrifuged at 10 000 × g for 10 min. Subsequently, the working solution was prepared according to the instructions of the kit and the reaction was carried out. Finally, the absorbance values at 532 nm (MDA) and 450 nm (SOD) were detected using a microplate reader. The experiment was performed with 5 biological replicates and 3 technical replicates.
Dual-Luciferase Reporter Assay
The binding sites of the miR-342-3p and HOXA11-AS were predicted using the lncRNASNP database. Subsequently, the wild-type sequence and its point-mutated sequence were respectively inserted into pmirGLO (Cat# E1330, Promega, USA). Using Lipofectamine 2000, the constructed reporter plasmid (100 ng) was co-transfected with miR-342-3p mimic or mimic NC (50 nM) into AC16 cells. After 48 h of transfection, the cells were collected and the luciferase activity was detected using the dual-luciferase reporter gene assay system (Cat# E1910, Promega, USA). Finally, the Renilla luciferase was used as the internal reference, and the firefly luciferase activity was normalized to calculate the relative luciferase activity.
Statistical Analysis
Statistical analysis was conducted using SPSS and GraphPad Prism software. All the data are derived from five independent biological experiments and are presented in the form of mean ± SD. Comparisons between groups were performed using Student's t-test or one-way ANOVA. When the ANOVA results indicated significant differences, Tukey post-hoc tests were employed for multiple comparisons. Statistical significance was defined as P < .05.
Results
Comparison of Baseline Clinical Data
The study enrolled 115 patients with CHF and 110 healthy individuals. The baseline clinical characteristics in two groups are shown in Table 1. The CHF group and the healthy control group showed no statistically significant differences in age, gender, BMI, smoking history, drinking history, hypertension, and lipid levels (P > .05). However, compared with the healthy control group, the BNP level in the CHF group was significantly increased, while the LVEF was significantly decreased (P < .001).
Comparison of Clinical Data Between Healthy Medical Examiners and Patients with CHF.
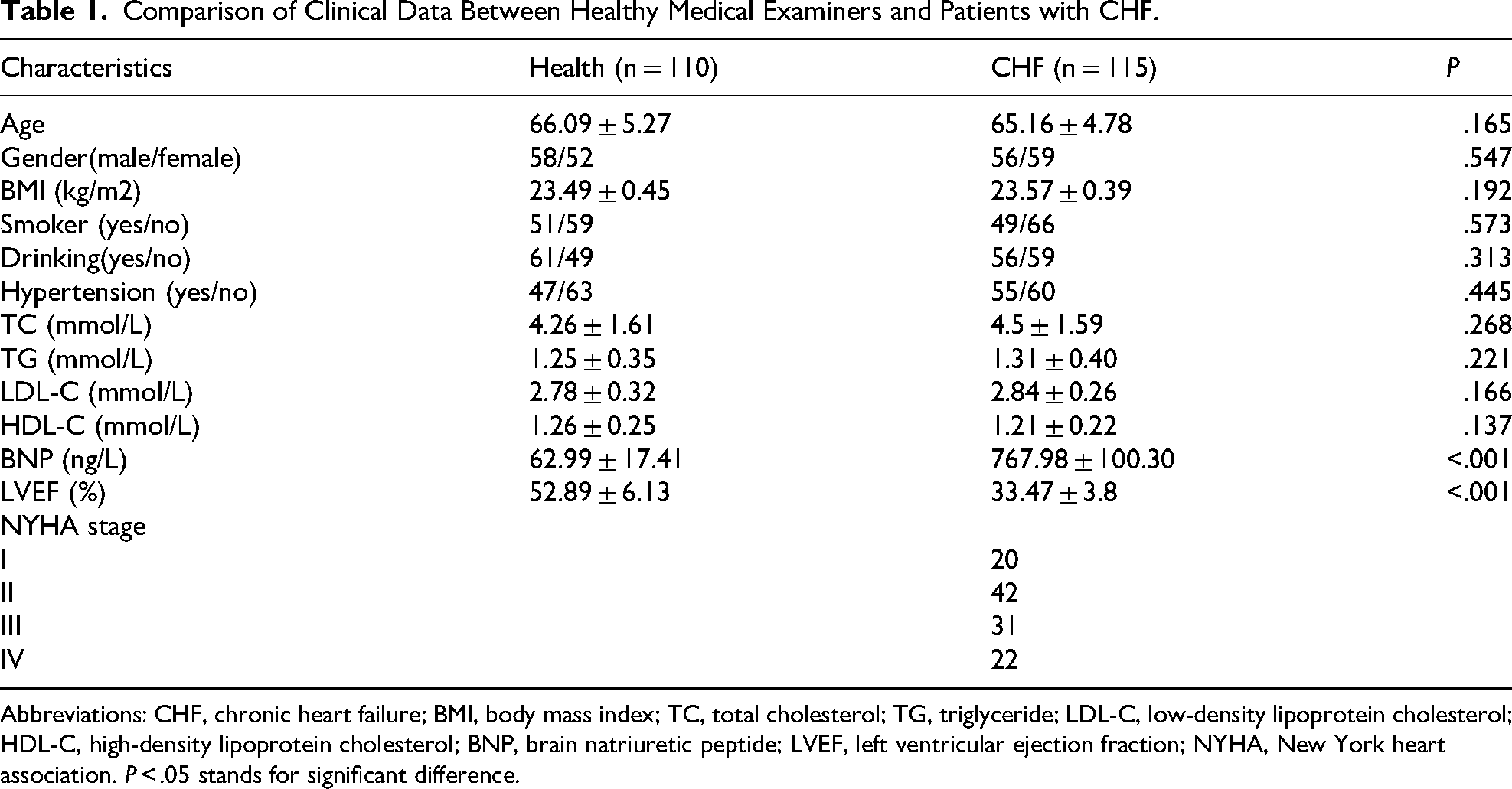
Abbreviations: CHF, chronic heart failure; BMI, body mass index; TC, total cholesterol; TG, triglyceride; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; BNP, brain natriuretic peptide; LVEF, left ventricular ejection fraction; NYHA, New York heart association. P < .05 stands for significant difference.
The Diagnostic Value of HOXA11-AS in CHF
We conducted RT-qPCR to detect HOXA11-AS in the serum. Compared with the healthy control, the expression level of HOXA11-AS was significantly increased in the serum of patients with CHF (P < .0001, Figure 1A). Additionally, the expression level of HOXA11-AS significantly increased as the NYHA cardiac function classification rises, indicating a correlation between it and the severity of heart failure (P < .01, Figure 1B). ROC curve analysis revealed that HOXA11-AS had good diagnostic value for CHF (AUC = 0.8821), with a sensitivity and specificity were 76.52% and 89.09%, respectively (Figure 1C).
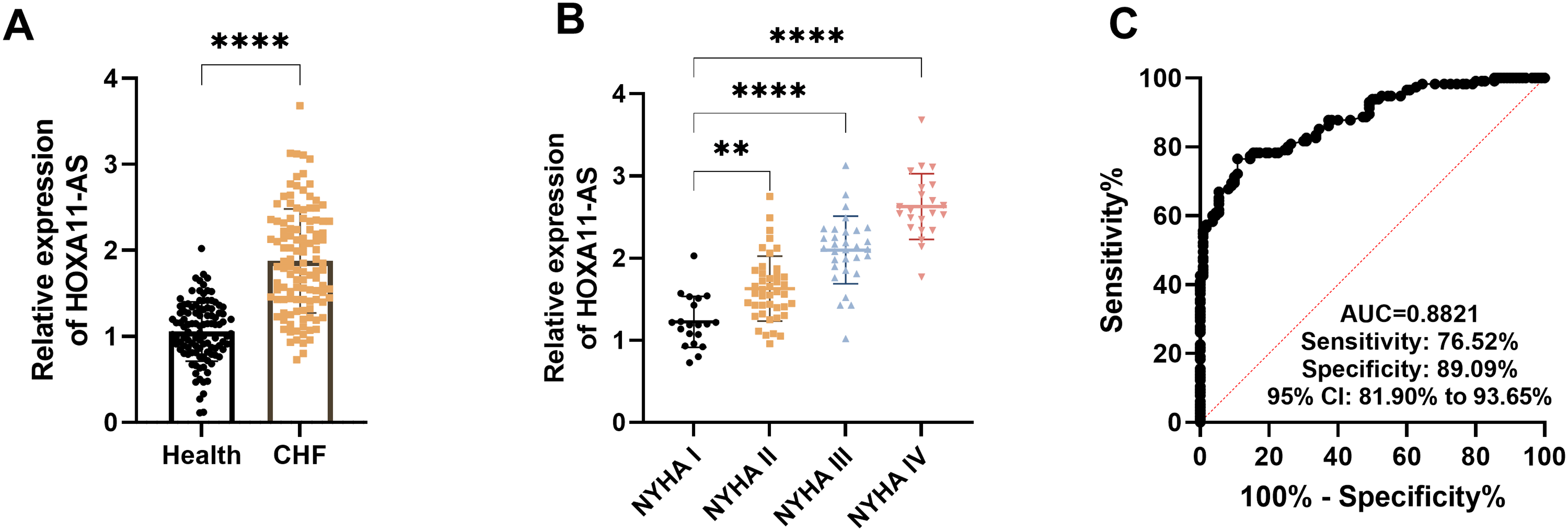
The diagnostic value of HOXA11-AS in chronic heart failure (CHF). A. The expression of HOXA11-AS in patients with CHF. B. The expression of HOXA11-AS in patients with different NYHA classification. C. The ROC curve indicates that HOXA11-AS has diagnostic value in CHF. ** P < .01, **** P < .0001.
HOXA11-AS Regulates the Cell Viability of Cardiomyocytes Induced by DOX
In order to simulate chronic heart failure in vitro, we cultivated the cardiomyocytes with DOX. DOX significantly reduced the viability of AC16 cells (P < .001, Figure 2A). Meanwhile, the expression of HOXA11-AS increased with the increase of DOX (P < .001, Figure 2B). To explore the role of HOXA11-AS in this model, we successfully knocked down its expression by siRNA (P < .05, Figure 2C). Functional experiments confirmed that knocking down the expression of HOXA11-AS could significantly alleviate the decrease in the viability of cardiomyocytes caused by DOX, and inhibit cell apoptosis (P < .001, Figure 2D and E).
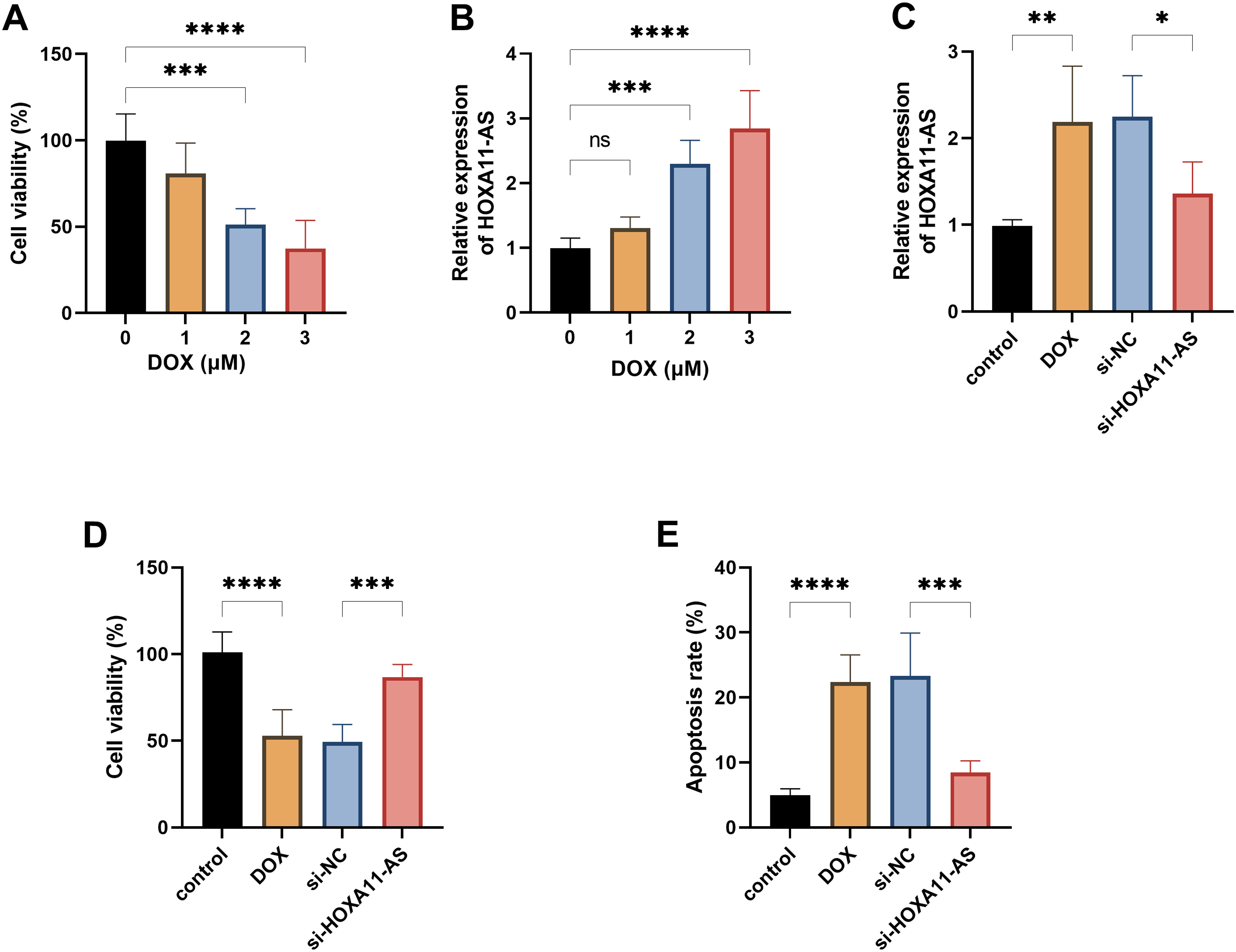
HOXA11-AS regulates the proliferation and apoptosis of cardiomyocytes induced by Doxorubicin (DOX). A. Cell viability of AC16 cells treated with different concentrations of DOX. B. The effects of DOX on the expression level of HOXA11-AS in AC16 cells. C. The expression of HOXA11-AS in AC16 cells after the transfection of siRNA. D. The effect of knocking down HOXA11-AS on cell proliferation. E. The effect of knocking down HOXA11-AS on cell apoptosis. * P < .05, ** P < .01, *** P < .001, **** P < .0001.
Knocking Down HOXA11-AS Inhibits Myocardial Injury, Inflammation and Oxidative Stress Caused by DOX
In order to deeply explore the mechanism by which HOXA11-AS causes myocardial injury, we measured a series of indicators that reflect myocardial damage. Compared with the si-NC group, knockdown of HOXA11-AS not only reduced the expression of myocardial injury markers such as ANP and BNP (P < .05, Figure 3A and B), but also inhibited the release of pro-inflammatory cytokines such as IL-1β and TNF-α (P < .001, Figure 3C and D). At the same time, this operation also significantly alleviated oxidative stress, which was confirmed by a decrease in MDA and the increase in SOD (P < .01, Figure 3E and F). These results indicate that HOXA11-AS can mediate the cardiomyocyte damage caused by DOX.
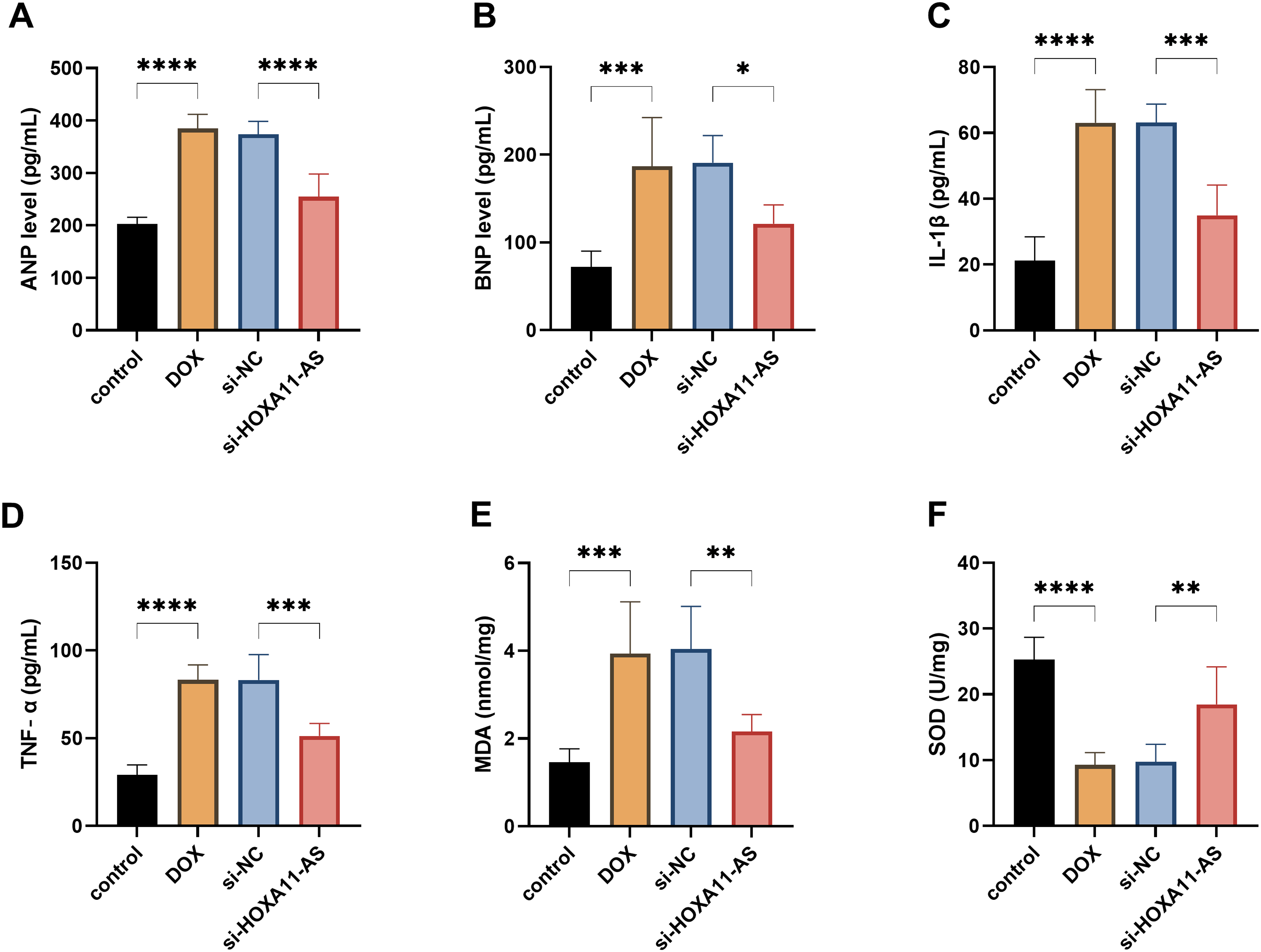
The downregulation of HOXA11-AS can alleviate the damage to cardiomyocytes caused by Doxorubicin (DOX). A–B. The effect of knocking down HOXA11-AS on the levels of cardiac injury markers. C–D. The effect of knocking down HOXA11-AS on the concentrations of inflammatory factors. E–F. The effect of knocking down HOXA11-AS on the levels of SOD and MDA. * P < .05, ** P < .01, *** P < .001, **** P < .0001.
miR-342-3p is the Downstream Target Gene of HOXA11-AS
Bioinformatics analysis indicated that there were potential interaction sites between HOXA11-AS and miR-342-3p (Figure 4A). The DLR assay demonstrated that miR-342-3p mimics could significantly inhibit the luciferase activity of the HOXA11-AS-WT, indicating a direct targeting binding relationship between the two (P < .001, Figure 4B). The expression of miR-342-3p was significantly downregulated in patients with CHF and AC16 cells treated with DOX (P < .001, Figure 4C and D). Correlation analysis showed that the expression levels of HOXA11-AS and miR-342-3p in the serum of patients with CHF were significantly negatively correlated (r = −0.6522, P < .0001, Figure 4E). Therefore, HOXA11-AS may participate in the pathogenesis of CHF by directly adsorbing miR-342-3p.
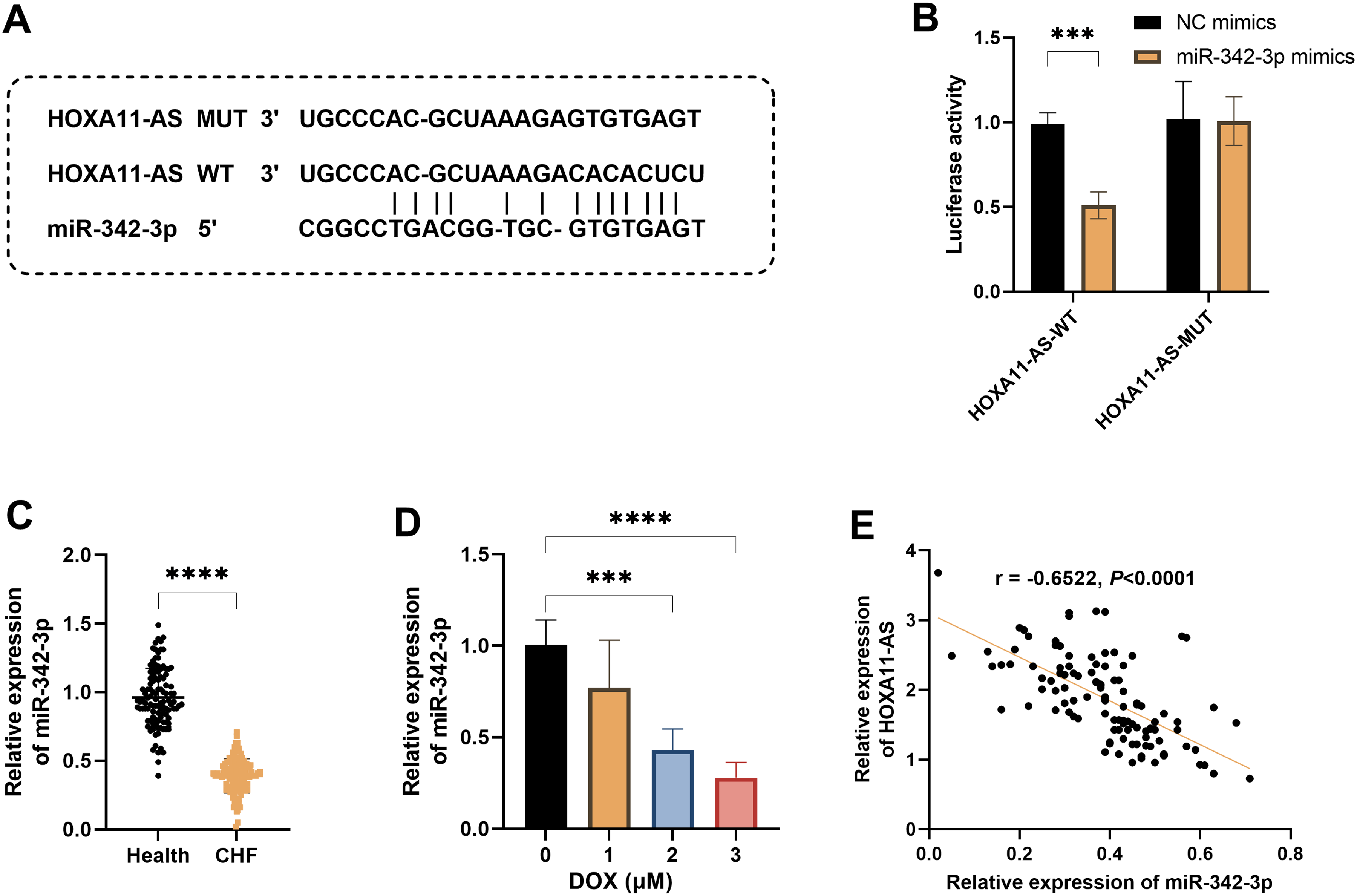
miR-342-3p is the downstream target gene of HOXA11-AS. A. The binding sites between miR-342-3p and HOXA11-AS were predicted by the lncRNASNP database. B. The dual-luciferase reporter gene assay revealed the targeting relationship between miR-342-3p and HOXA11-AS. C. The expression level of miR-342-3p in patients with chronic heart failure (CHF) and healthy controls. D. The expression levels of miR-342-3p in AC16 cells treated with different concentrations of Doxorubicin (DOX). E. The correlation between the expression of miR-342-3p and HOXA11-AS in patients with CHF. *** P < .001, **** P < .0001.
The miR-342-3p Inhibitor Weakened the Protective Effect of Silencing HOXA11-AS on Cardiomyocytes
In order to verify whether HOXA11-AS exerts its effect through miR-342-3p, we simultaneously transfected si-HOXA11-AS and miR-342-3p inhibitors. The results showed that the miR-342-3p inhibitor could significantly reverse the cell protective effect caused by the knockdown of HOXA11-AS. Specifically, the inhibition of miR-342-3p weakened the cell proliferation ability, exacerbated cell apoptosis, and simultaneously promoted the expression of cardiac injury markers and pro-inflammatory factors (P < .05, Figure 5A–F). Moreover, the level of oxidative stress also deteriorated, manifested by an increase in MDA and a decrease in SOD (P < .05, Figure 5G and H).
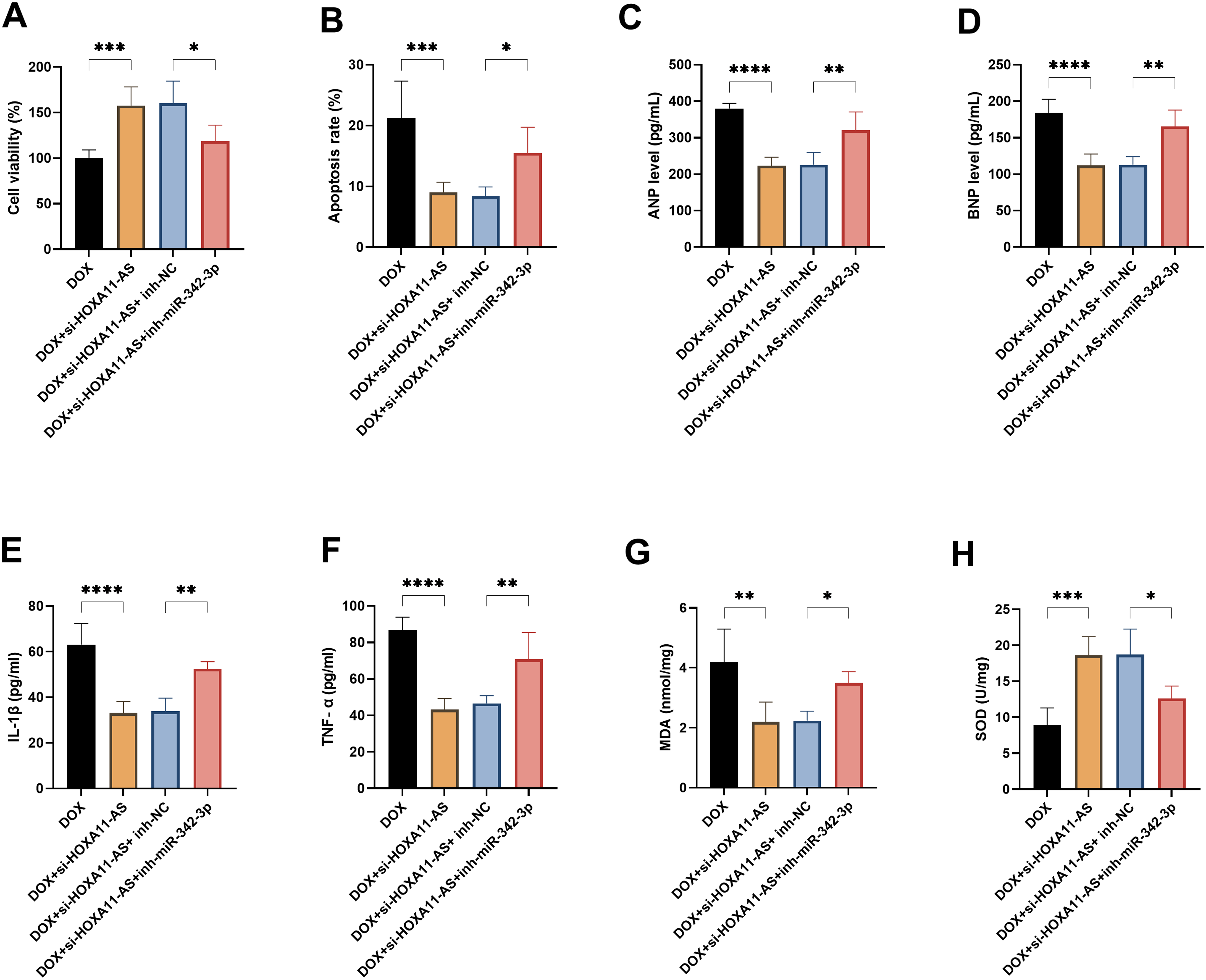
The miR-342-3p inhibitor reversed the protective effect of HOXA11-AS silencing on cardiomyocytes. A. Measurement of cell proliferation activity. B. Cell apoptosis rate. C–D. Concentrations of myocardial injury markers. E–F. The concentration of inflammatory factors. G-H. The content of MDA and the activity of SOD. * P < .05, ** P < .01, *** P < .001, **** P < .0001.
Discussion
CHF is a complex clinical syndrome, and its pathogenesis involves multiple aspects, including myocardial injury, inflammatory response, oxidative stress, and fibrotic remodeling of the heart.20,21 DOX is an anthracycline chemotherapy drug that can cause cardiac toxicity through mechanisms such as inducing oxidative stress, mitochondrial dysfunction, inflammatory response, and apoptosis. These pathological features are highly consistent with the key pathogenesis of CHF.22,23 Therefore, in this study, we used DOX to induce the AC16 cells to establish an in vitro model of CHF. Based on this, we explored the potential mechanism of HOXA11-AS in CHF.
LncRNA plays diverse multiple regulatory roles in the occurrence and development of CHF. They not only participate in pathological processes such as myocardial remodeling and fibrosis, but also show potential as novel diagnostic and prognostic markers.24–26 For instance, HOTAIR can alleviate cardiac injury by regulating miR-30a-5p, thereby improving cardiac injury in CHF 27 ; SMILR participates in angiogenesis by influencing miR-10b-3p. 28 Inflammation is an important stage in the onset of CHF, and HOXA11-AS has been proven to promote inflammatory responses in various diseases such as nephritis and neuroinflammation.29–31 This indicates that it may be involved in the pathological development process of CHF through inflammatory mechanisms.
In this study, we first discovered that the level of HOXA11-AS in the serum of patients with CHF was significantly upregulated, and this elevation was closely associated with the degree of heart failure. Further analysis confirmed that HOXA11-AS has a good diagnostic ability for CHF, suggesting that it may become a new biomarker. At the diagnostic level, HOXA11-AS can be combined with existing markers to construct a more accurate diagnostic model. At the management level, these dynamic changes can be utilized to assess the progression of the disease, the effectiveness of the treatment, and the risk of readmission. Furthermore, from the perspective of developing therapeutic targets, specific inhibition of HOXA11-AS may provide new ideas for targeted treatment of heart failure. To further explore the specific role of HOXA11-AS in myocardial injury, we established a myocardial cell injury model. The experimental results showed that knocking down HOXA11-AS could significantly improve cell viability, inhibit cell apoptosis, and reduce the expression of myocardial injury markers. Moreover, knocking down HOXA11-AS also greatly inhibited the release of inflammatory factors and the level of oxidative stress. This indicates that by regulating the inflammatory and oxidative stress pathways, HOXA11-AS plays a role in promoting myocardial injury.
The research on miR-342-3p was initially conducted in the field of oncology, and it has been confirmed that it plays a significant role in the pathogenesis of various malignant tumors.32–34 In recent years, with the continuous advancement of research, relevant studies have shown that miR-342-3p may be a potential factor in the pathogenesis of chronic heart failure. There is a direct targeting relationship between HOXA11-AS and miR-342-3p. miR-342-3p was significantly reduced in patients with CHF and DOX-treated cells, and it was negatively correlated with HOXA11-AS. More importantly, inhibiting miR-342-3p can reverse the cell protective effect caused by the knocking down of HOXA11-AS. This indicates that in CHF, HOXA11-AS mainly exacerbates the destruction of cardiomyocytes by inhibiting miR-342-3p.
Based on the existing literature and bioinformatics analysis, we hypothesize that miR-342-3p may exert myocardial protective effects through multiple mechanisms. Specifically, in terms of the death of cardiomyocytes, miR-342-3p can reduce cell apoptosis by directly act on the transcription factors SOX6 and TFEB. 16 In terms of inflammation, bioinformatics analysis predicts that it may target key inflammatory signaling molecules such as TLR4 and inhibiting the activation of NF-κB pathways. Moreover, given the crucial role of myocardial fibrosis in heart failure, miR-342-3p may also inhibit the activation of fibroblasts and the deposition of extracellular matrix by targeting TGF-β/Smad pathway. 35 In summary, the HOXA11-AS/miR-342-3p axis may be involved in the development of CHF by influencing various pathological processes such as apoptosis, inflammation, and fibrosis. Future research need to combine experimental verification with biological network analysis in order to systematically and clearly identify the specific downstream target genes and regulatory networks of this axis.
In conclusion, this study has demonstrated that HOXA11-AS has a significant diagnostic value for CHF. Furthermore, HOXA11-AS may regulate myocardial injury, inflammatory response, and oxidative stress by targeting miR-342-3p, and thereby participate in the pathological process of CHF.
Limitations
Our research was mainly conducted in cardiomyocytes. Although this method enabled us to analyze the direct effects of the HOXA11-AS/miR-342-3p axis on cardiomyocytes, it failed to fully demonstrate the complex multicellular environment in the failing heart. The progression of CHF involves interactions among various cell types, such as cardiac fibroblasts, endothelial cells, and immune cells, etc Further studies are needed in more cell models and in vivo models to more comprehensively reveal the regulatory mechanism of this molecular axis in CHF.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Multicenter Randomized Controlled Trial of Integrated Primary Care Intervention for Weight Optimization in Patients with Four Highs: A Multi-Target Collaborative Management Approach (No. GWJJMB202510010106) and Establishing and Implementing a Community-Based Smart Management System for Tiered Collaborative Prevention and Control Across the Cardiovascular Event Chain in Elderly Patients with Coexisting Hypertension, Hyperlipidemia, and Hyperglycemia (No. 2023YFC3605005).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.