
Abstract
Objective:
To characterize the clinical and genomic features of vancomycin-resistant enterococci (VRE) in a tertiary hospital in Huizhou and identify risk factors to inform local infection control.
Methods:
A retrospective study included 58 VRE and 25 vancomycin-susceptible Enterococci (VSE) strains (August 2023–May 2025). Clinical data and antimicrobial susceptibility were analyzed; whole-genome sequencing (WGS) was performed on 54 VRE strains.
Results:
Midstream urine was the primary VRE-positive specimen. ICU admission, polyantibiotic use (≥3 agents), and urinary catheterization were key risk factors for VRE. All VRE isolates were Enterococcus faecium and showed a predominantly clonal population structure, dominated by CC17/ST80 (68.8%) and CC2/ST106 (64.6%) under the two multilocus sequence typing schemes; five novel STs were ultimately identified in the latter scheme. VRE was universally resistant to ampicillin, with high resistance to penicillin, levofloxacin, and teicoplanin, while linezolid and tigecycline remained effective. Genotypically, 94.8% carried vanA, 100% carried virulence gene esp, and aminoglycoside and macrolide resistance genes were prevalent. A unique VRE strain (VRE48) showed resistance without canonical van genes, harboring a Ddl Ser210Tyr mutation.
Keywords
Introduction
Enterococci are usually colonized in the intestines of humans and animals and are part of the gastrointestinal flora of healthy individuals. 1 Although enterococci are intestinal commensals, they are also common nosocomial pathogens that can cause urinary tract infections, bacteremia, endocarditis, meningitis, and other infections in immunocompromised patients. 2 Enterococci are now the second most commonly isolated Gram-positive pathogens in hospitals, after Staphylococcus aureus, a trend linked to the increased use of invasive procedures, hormones, and antimicrobials. Their intrinsic and acquired resistance further limits treatment options,3,4 especially the prevalence of vancomycin-resistant enterococci (VRE), which has become a core challenge in infection control and treatment. 5 Glycopeptide antibiotics represented by vancomycin inhibit transglycosylation and tran-speptidation reactions by specifically binding to the D-alanine-D-alanine (D-Ala-D-Ala) site at the terminal of bacterial cell wall peptidoglycan precursors, ultimately blocking cell wall synthesis. 6 To date, at least nine glycopeptide resistance genotypes (vanA, vanB, vanC, vanD, vanE, vanG, vanL, vanM, vanN) have been identified, with vanA and vanB being the most common. These genes encode enzymes that produce low-affinity peptidoglycan precursors, altering vancomycins target site and preventing binding, thereby conferring resistance to vancomycin and other glycopeptides.7,8
Since the first report of VRE in 1988, the detection rate of such bacteria has increased year by year. 9 VRE has shown an increasing trend in most countries around the world, causing outbreaks and severe infections in Europe, the Americas, and Australia.10,11 According to data from the China Antimicrobial Surveillance Network (http://www.carss.cn/), the prevalence of VRE in China has been very low (average 5%) over the past few decades. Although the incidence is low, compared with VSE infections, VRE infections are associated with increased morbidity, mortality, medical costs, and length of hospital stay. 12 However, VRE prevalence in China shows localized outbreaks. A multicenter study found that VRE rates in Guangdong Province have risen from 5% before 2021 to 20%–50% in 2023, while remaining stable elsewhere. ST17 and ST78 have been replaced by the emerging ST80, which forms a distinct lineage genetically distant from ST80 VRE in other countries, suggesting a regional ST80 outbreak.13,14
VRE prevalence varies globally, mainly due to differences in medical resources and antibiotic use. Most studies focus on large cities, and domestic data on VRE clinical phenotypes and genomic characteristics remain limited, with no reports from Huizhou. As a key city in the Guangdong-Hong Kong-Macao Greater Bay Area, Huizhou has high population mobility and antibiotic pressure, highlighting the need for systematic VRE surveillance. Whole-Genome Sequencing(WGS) data can clarify genetic features and transmission dynamics, support early detection and rapid response to outbreaks, and enable timely interventions by monitoring major and emerging STs.15,16
A total of 58 VRE and 25 VSE strains were collected from a tertiary hospital in Huizhou between August 2023 and May 2025. Clinical characteristics and antimicrobial susceptibility were compared, and genomic DNA extraction and WGS were performed on 58 VRE strains to support VRE prevention and control.
Materials and Methods
Sample collection and identification of Enterococcus faecium
From August 2023 to May 2025, VRE strains were collected from inpatients and outpatients at Huizhou Central People’s Hospital. To minimize intergroup confounding, only contemporaneous Bloodstream Infection-derived (BSI-derived) VSE strains were included for clinical characteristic comparison with VRE, controlling potential confounding factors between the two groups. Isolates were cultured on Columbia blood agar (5% sheep blood) and identified by Bruker Microflex Linear Benchtop/Smart High (LT/SH) mass spectrometry. This retrospective study was approved by the Clinical Research Ethics Committee of Huizhou Central People’s Hospital (approval number: ky112025179).
Antimicrobial susceptibility testing and collection of clinical information
Antimicrobial susceptibility testing (quantitative, routine) was performed on mass spectrometry-identified Enterococcus strains using the VITEK 2-Compact system to determine the minimum inhibitory concentration (MIC) of tested agents: penicillin, ampicillin, high-level gentamicin, levofloxacin, erythromycin, linezolid, teicoplanin, vancomycin, and tigecycline. Isolates with a vancomycin MIC ≥32 μg/mL were defined as VRE, and those with a vancomycin MIC ≤4 μg/mL were classified as VSE. Susceptibility interpretations for tigecycline followed the criteria established by the Food and Drug Administration, while those for the remaining agents adhered to the standards set forth by the Clinical and Laboratory Standards Institute (CLSI, https://clsi.org/). Collected patient information included demographics, underlying diseases, invasive procedures, prior antibiotics and antimicrobial susceptibility profiles.
Extraction and sequencing of Whole-Genome sequences of VRE isolates
Genomic DNA was extracted using the STE method, assessed for purity and integrity by agarose gel electrophoresis, and quantified by Qubit. Libraries were constructed with the NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA) via end repair, A-tailing, adapter ligation, purification, and PCR amplification. Validated libraries were sequenced on an Illumina NovaSeq platform with PE150 reads.
WGS analysis
Raw reads were filtered with Trimmomatic v0.39 17 and de novo assembly was performed using SPAdes v3.14.0, 18 with quality assessed by QUAST v5.0.2 19 to obtain genome size, number of contigs, and N50 values. Genome completeness was evaluated using BUSCO v6.0.020 with the bacteria_odb10 lineage dataset. Mean sequencing coverage was calculated from FASTA headers as a weighted average of contig lengths and coverage values. Contigs were taxonomically classified with Kraken2 v2.1.3, 20 and non-Enterococcus faecium sequences were removed using seqkit v2.10.0. 21 Core SNPs were identified by Snippy v4.6.0 against reference genome SRR24 (NZ_CP038996), and a maximum-likelihood phylogenetic tree was constructed with IQ-TREE v3.0.1, 22 and visualized in iTOL v7. 23 Two existing schemes were employed for multilocus sequence typing (MLST): one was the original scheme proposed by Homan et al., 24 and the other was the updated scheme described by Bezdicek et al. 25 Allele profiles and sequence types (STs) were determined with the assistance of the PubMLST database (https://pubmlst.org/). 26
chewBBACA v3.4.1 was used for gene-by-gene typing to compare whole-genome allelic variation among Enterococcus faecium strains. 27 A species-specific training file was generated from the reference genome SRR24 (NZ_CP038996) using Prodigal v2.6.3 to build a wgMLST scheme 28 from which a cgMLST scheme was derived by retaining loci present in all genomes. cgMLST allelic profiles were visualized using PHYLOViZ Online web platform (https://online.phyloviz.net), 29 with minimum spanning trees (MSTs) and population structure plots constructed based on allelic distances. Resistance and virulence genes were identified using ABRicate v1.0.1 with the ResFinder, 30 VFDB, 31 and CARD 32 databases. Hierarchical clustering of resistance gene profiles was performed using Euclidean distance and complete linkage, and heatmaps were generated with Chiplot. Amino acid sequences of the D-Ala-D-Ala ligase (Ddl) were extracted from the selected isolates and aligned using MAFFT v7.525 with default parameters. 33 The resulting multiple sequence alignment was visualized using ESPript v3.0. 34
Statistical methods
Statistical analysis of data was performed using SPSS 29.0. The test method was selected based on the data type and distribution characteristics: the Pearson’s chi-square test was used when the expected frequency of all cells was ≥5 and the total sample size was ≥40.
Results
Analysis of clinical infection characteristics of VRE
Of the 58 VRE isolates from inpatients, 4 were from outpatients with incomplete clinical data, leaving 54 cases for analysis. VRE was most commonly isolated from midstream urine (29/54, 54%). Among the remaining 25 VRE isolates, peritoneal fluid and blood each accounted for 11% (6/54), wound secretion and sputum each accounted for 7% (4/54), bile accounted for 5% (3/54), and pleural effusion and indwelling catheter each accounted for 2% (1/54). Thirty-one patients had been admitted to the intensive care unit (ICU), a higher proportion than non-ICU departments. No significant difference in VRE detection was observed between males and females (p > 0.05). ICU patients had significantly higher rates of lung disease, central nervous system disease, and invasive procedures compared with non-ICU patients (p < 0.05) (Table 1).
Comparison of Underlying Diseases and Invasive Procedures between ICU and Non-ICU Groups in VRE Patients
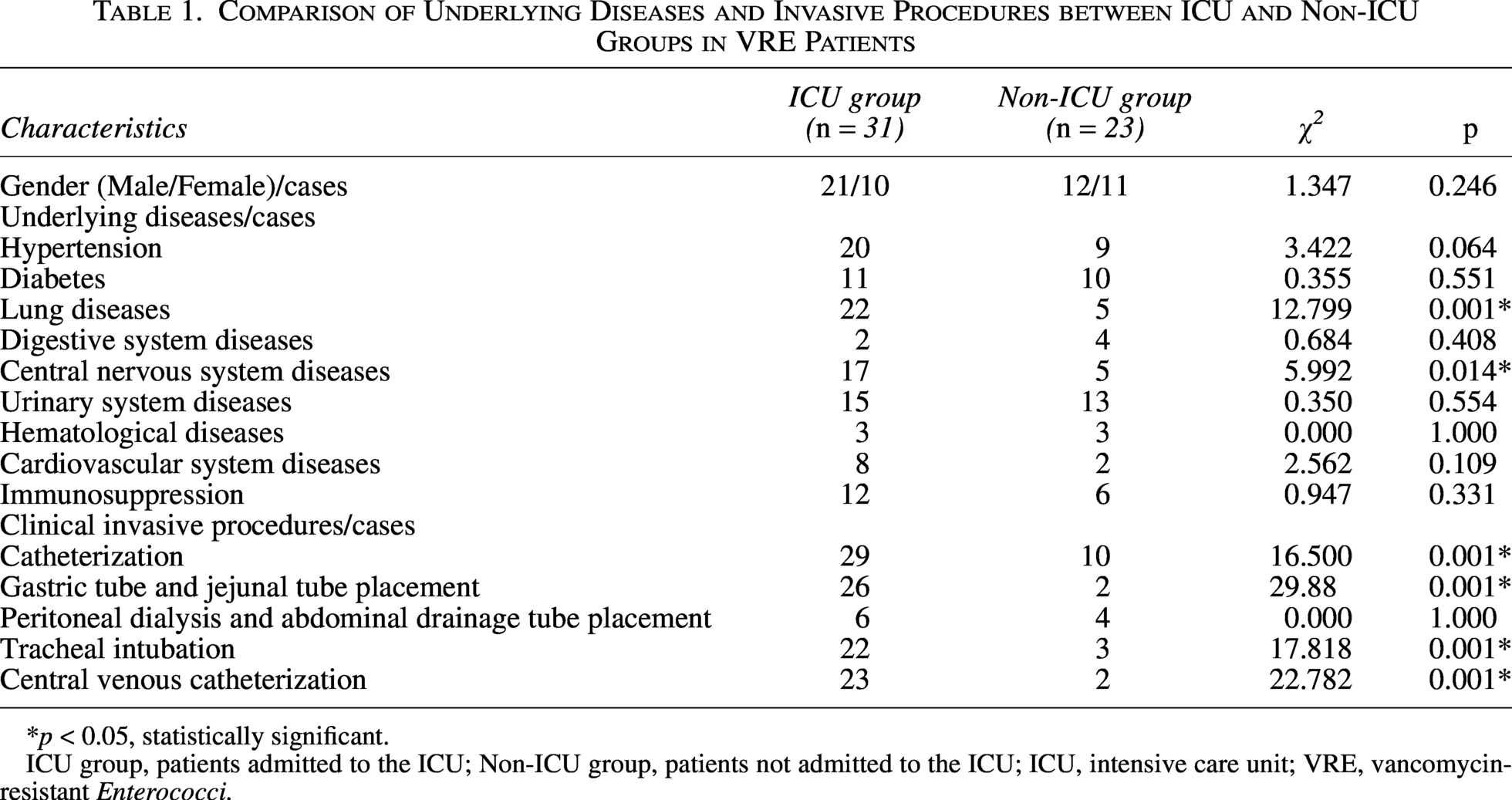
*p < 0.05, statistically significant.
ICU group, patients admitted to the ICU; Non-ICU group, patients not admitted to the ICU; ICU, intensive care unit; VRE, vancomycin-resistant Enterococci.
Comparative analysis of infection characteristics and antimicrobial susceptibility between VRE and VSE
Patients in both the VRE and VSE groups were predominantly male. Hypertension, diabetes mellitus, and pulmonary diseases were the most common underlying conditions, and urinary catheterization and central venous catheterization were the main invasive procedures in both groups. Compared with the VSE group, the VRE group showed statistically significant differences in male proportion, central nervous system diseases, and urinary catheterization rate (p < 0.05). Regarding antimicrobial exposure before pathogen detection, β-lactams were the most frequently used agents in both groups. The VRE group had significantly higher rates of vancomycin use and exposure to ≥3 antibiotics prior to isolation than the VSE group (p < 0.05) (Table 2). Multivariate Logistic regression analysis (Table 3) revealed that urinary catheterization (OR = 4.482, 95%CI: 1.266–15.870, p = 0.020) and use of ≥3 antibiotics before detection (OR = 3.768, 95%CI: 1.032–13.753, p = 0.045) were independent risk factors for VRE infection, whereas male sex (OR = 0.175, 95%CI: 0.038–0.806, p = 0.025) was a protective factor. Over 90% of VRE isolates were resistant to penicillin, levofloxacin, and teicoplanin, with complete resistance to ampicillin. Non-susceptibility to linezolid and tigecycline was low (0% and 1.9%, respectively). Compared with VSE, VRE showed significantly higher resistance to ampicillin, gentamicin, levofloxacin, erythromycin, and teicoplanin (p < 0.05) (Table 4).
Comparison of Clinical Characteristics between VRE and VSE Groups
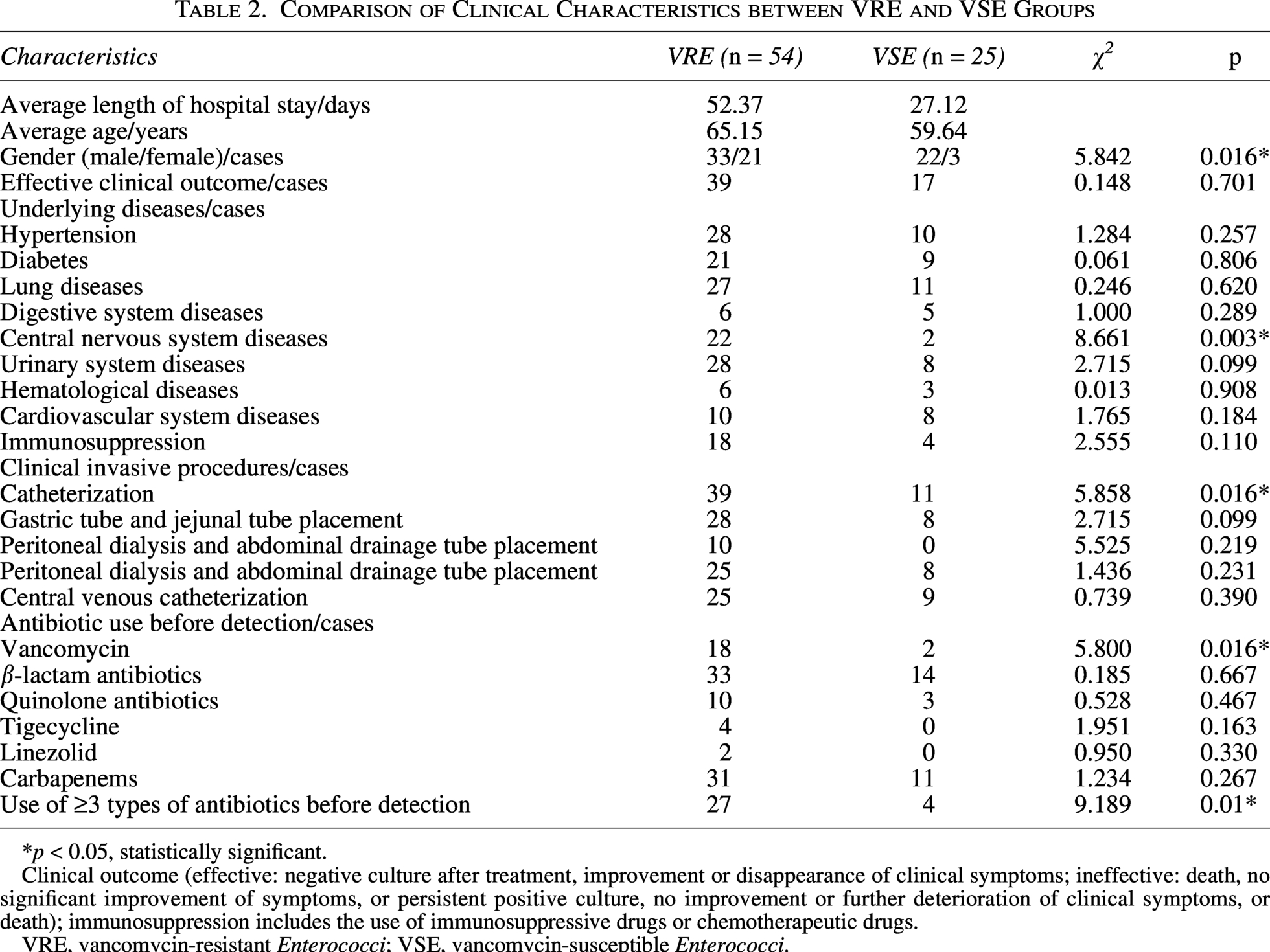
*p < 0.05, statistically significant.
Clinical outcome (effective: negative culture after treatment, improvement or disappearance of clinical symptoms; ineffective: death, no significant improvement of symptoms, or persistent positive culture, no improvement or further deterioration of clinical symptoms, or death); immunosuppression includes the use of immunosuppressive drugs or chemotherapeutic drugs.
VRE, vancomycin-resistant Enterococci; VSE, vancomycin-susceptible Enterococci.
Multivariate Logistic Regression Analysis of Clinical Characteristics in Patients Infected with VRE and VSE
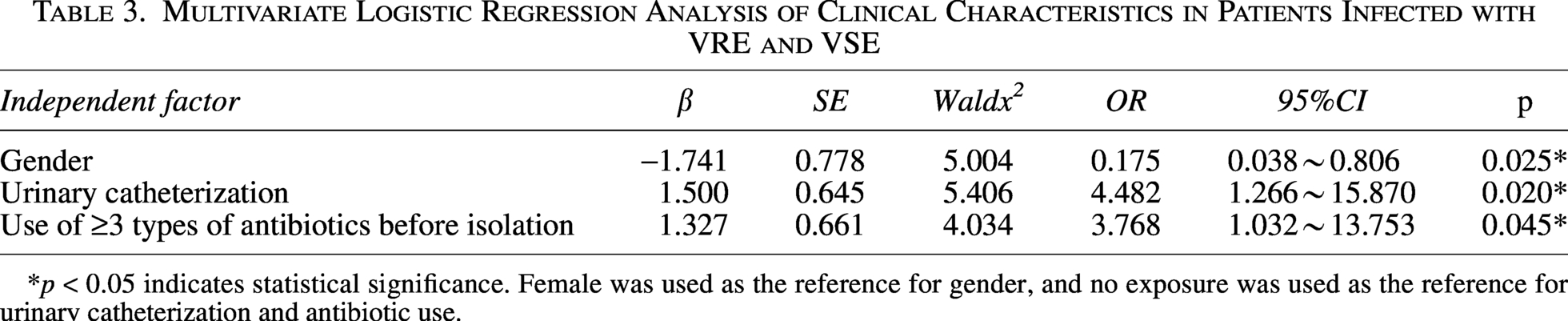
*p < 0.05 indicates statistical significance. Female was used as the reference for gender, and no exposure was used as the reference for urinary catheterization and antibiotic use.
Comparison of Drug Resistance between VRE and VSE Groups
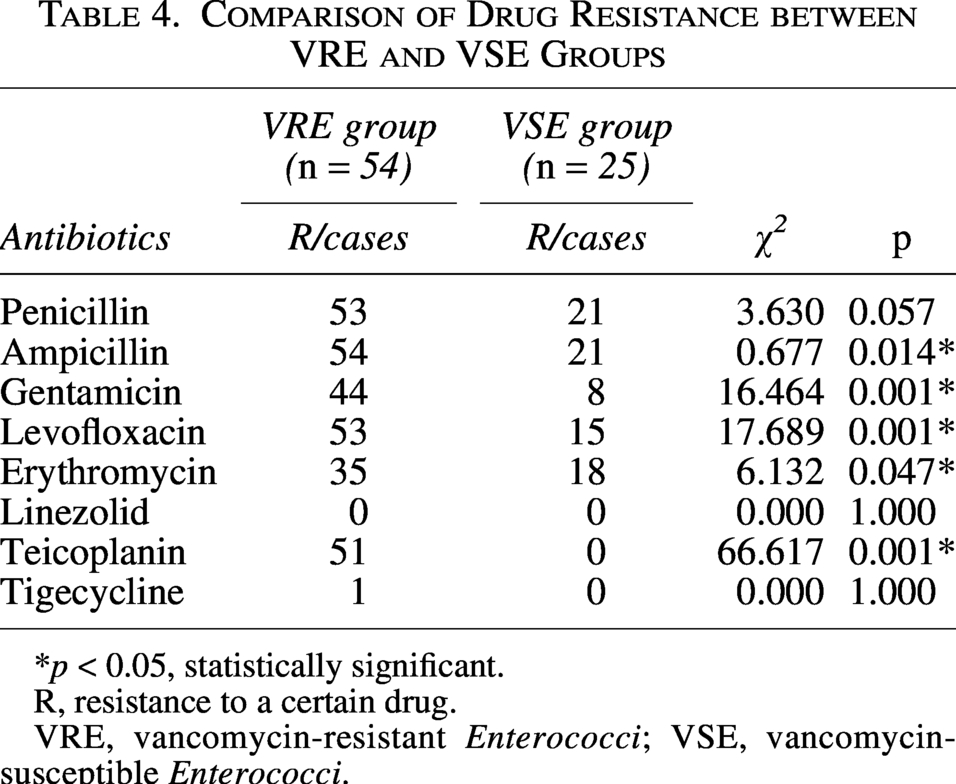
*p < 0.05, statistically significant.
R, resistance to a certain drug.
VRE, vancomycin-resistant Enterococci; VSE, vancomycin-susceptible Enterococci.
Phylogenetic tree analysis of VRE
In this study, we constructed a maximum-likelihood phylogenetic tree based on core single nucleotide polymorphism (SNP) to place our VRE strains in a global context (Fig. 1), excluding six distantly related isolates for clarity. Most local strains formed a distinct, well-separated cluster, highlighting a unique genetic background and evolutionary trajectory of VRE circulating in this region.
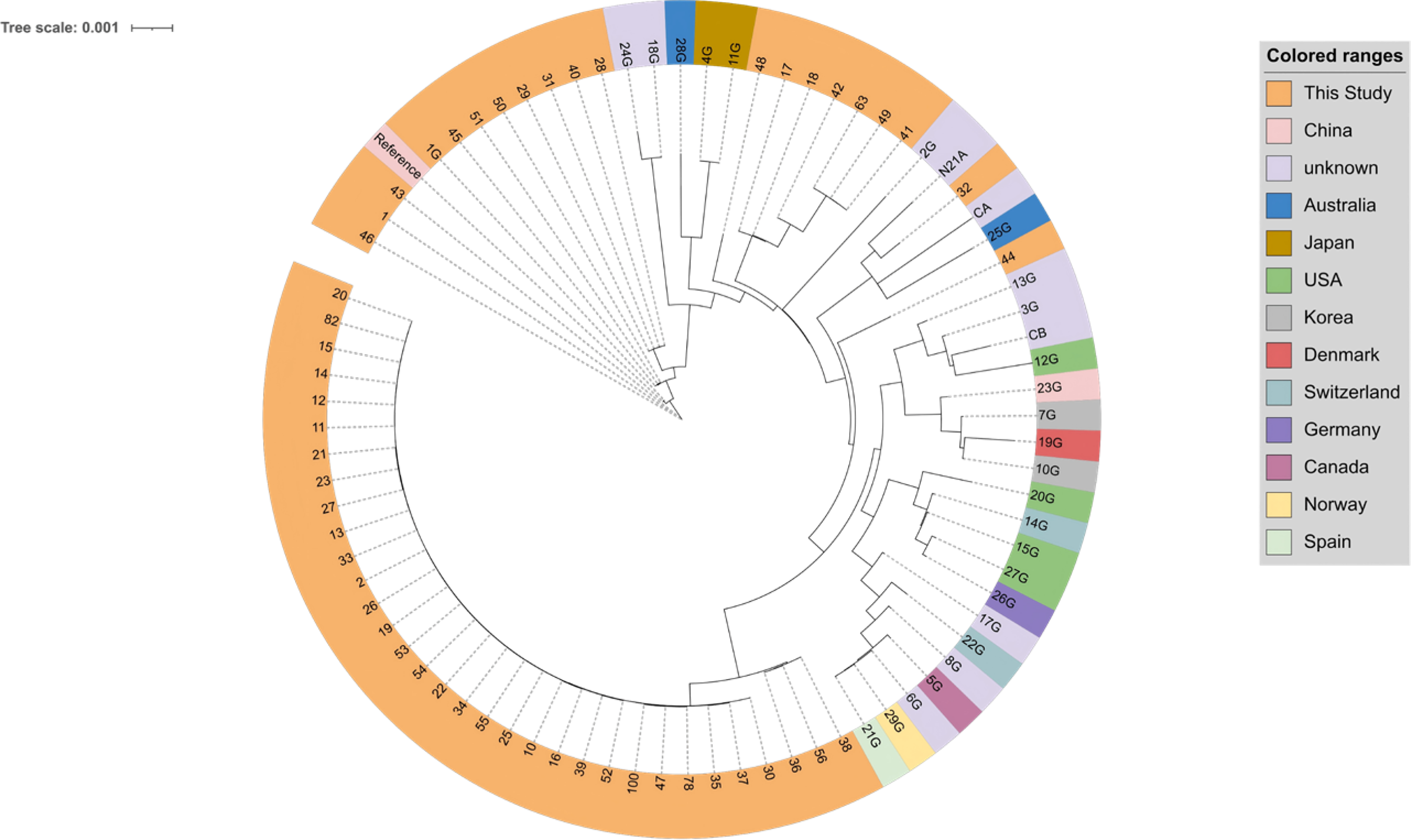
Global phylogenetic analysis of VRE. Phylogenetic tree illustrates the genetic relationships between VRE strains isolated in this study (orange branches, labeled with numerical IDs) and global reference strains (branches labeled with “G” suffix, color-coded by country of origin, as indicated in the legend); The tree scale (0.001) indicates the genetic variation degree, representing the number of core SNP differences per site; SNP, single nucleotide polymorphism.
Strain 48 (study isolate) formed a basal branch, while strain 28G (Australia) represented an intermediate branch leading to the formation of a sister pair of strains 4G and 11G (Japan) within the same sub-lineage. A separate sub-lineage comprised three parallel branches represented by strains 32 and 21A (unknown origin), strain CA (unknown origin), and strain 25G (Australia), with strain 44 located on a more divergent branch.
Assembly quality metrics including mean coverage depth, N50, genome completeness, and number of contigs are shown in Supplementary Table S1.
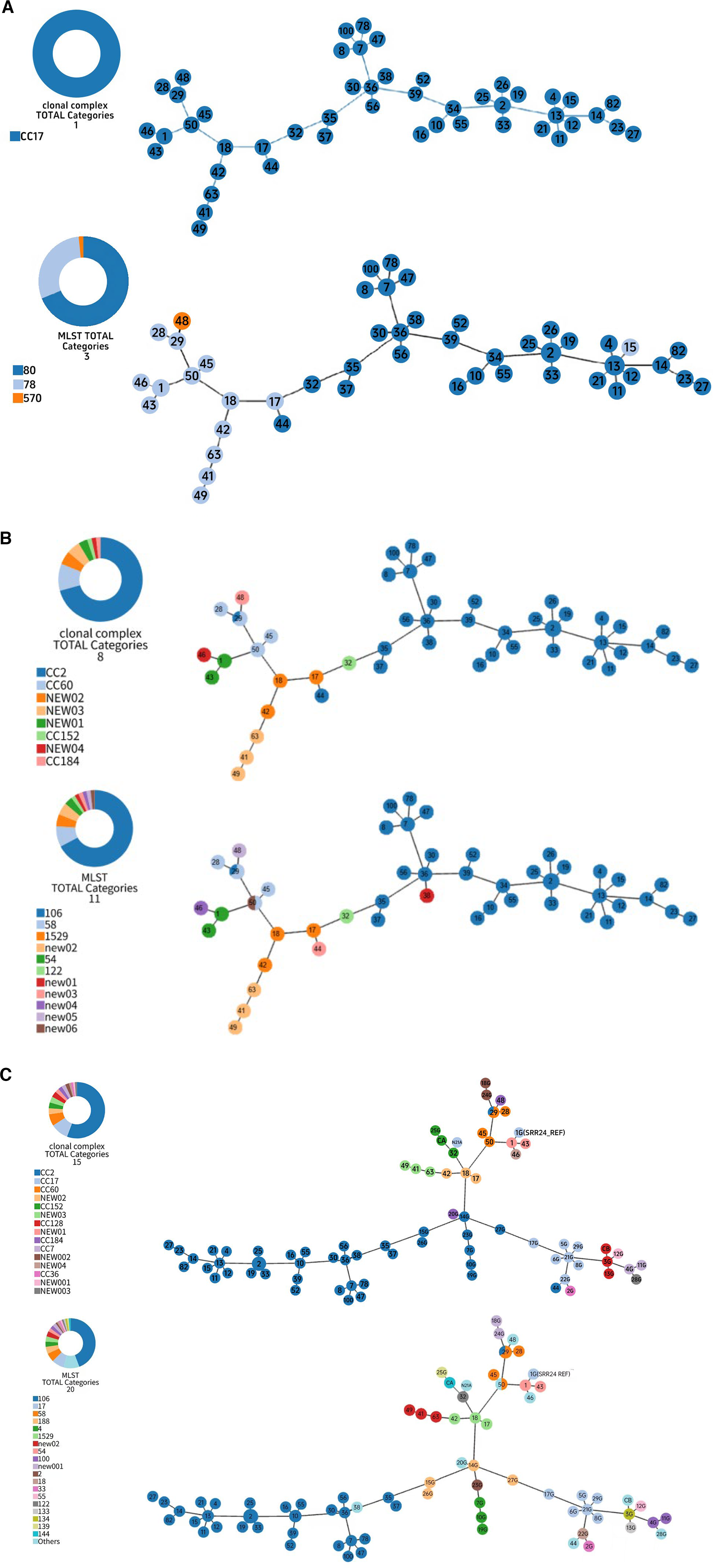
WgMLST molecular typing of VRE
An MST was constructed to visualize inter-strain genetic relationships, with phylogeny annotated by classical MLST 24 and extended MLST. 25 Of 58 initial isolates, 48 were retained in the MST after excluding those with excessive missing cgMLST loci. Classical MLST assigned all 48 VRE isolates to CC17, with ST80 as the dominant type (33/48, 68.8%), followed by ST78 (14/48, 29.1%) and ST570 (1/48, 2.1%) (Fig. 2a). Extended MLST yielded higher resolution, identifying 8 CCs (dominant CC2, 33/48, 68.8%) and 11 STs (dominant ST106, 31/48, 64.6%), with five novel STs (ST1588 and ST1591–ST1594) assigned in the PubMLST database. For global contextualization, the prevalent ST106/CC2 did not cluster directly with global reference strains, indicating an independent local transmission chain in Huizhou. Several study strains linked directly to the MST backbone and shared a genetic hub with references from other Chinese cities, the USA, Switzerland and Australia, implying potential common ancestry or cross-regional spread (Fig. 2c).
Correlation analysis of resistance genes and virulence genes in VRE
Of the 58 VRE strains, 94.8% carried vanA, 1.7% carried vanB, and 18.9% carried vanM. All strains (100%) harbored the esp(enterococcal surface protein) virulence gene, while gelE(gelatinase gene), asa1 (aggregation substance gene 1), and hyl (hyaluronidase gene) were not detected. High rates of aminoglycoside resistance genes, including aph (aminoglycoside phosphotransferase), aac (aminoglycoside acetyltransferase), and ant (aminoglycoside nucleotidyltransferase), as well as the macrolide resistance gene ermB (erythromycin ribosome methylase gene B), were also observed. Strains within the same phylogenetic cluster showed nearly identical profiles of van genes and multidrug resistance genes (Fig. 3).
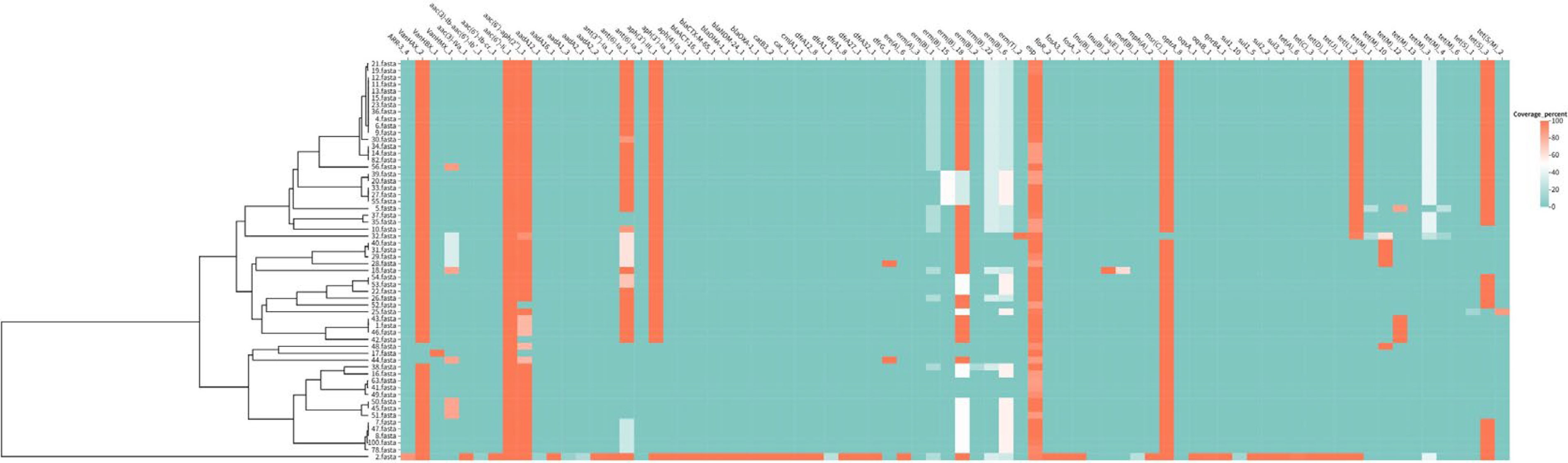
Correlation analysis of resistance genes and virulence genes. Heatmap showing the distribution of resistance genes (vanA, vanB, ermB, aac, aph, ant) and virulence genes esp in 54 VRE strains. The vertical axis represents VRE strains (grouped by cgMLST clusters), and the horizontal axis represents target genes. Red indicates the presence of the gene, and blue indicates the absence of the gene. Correlation matrix of resistance genes and virulence genes. The color intensity represents the correlation coefficient (range: −1 to 1), with dark red indicating a strong positive correlation and dark blue indicating a strong negative correlation.
Nonclassical resistance mechanisms in VRE
Vancomycin-resistant strains with canonical van clusters had identical Ddl sequences. However, strain 48(VRE48) in this study showed phenotypic vancomycin resistance without detectable van genes. For exploring potential related factors, the Ddl amino acid sequence of VRE48 was compared with three closely related vancomycin-susceptible E. faecium from the same hospital (Fig. 4). Multiple sequence alignment identified only two Ddl amino acid variations between VRE48 and susceptible strains FM111, FM91, FM80; a unique Ser210Tyr substitution at position 210 was present in VRE48, with a conserved serine at this site in all susceptible strains (Fig. 5).
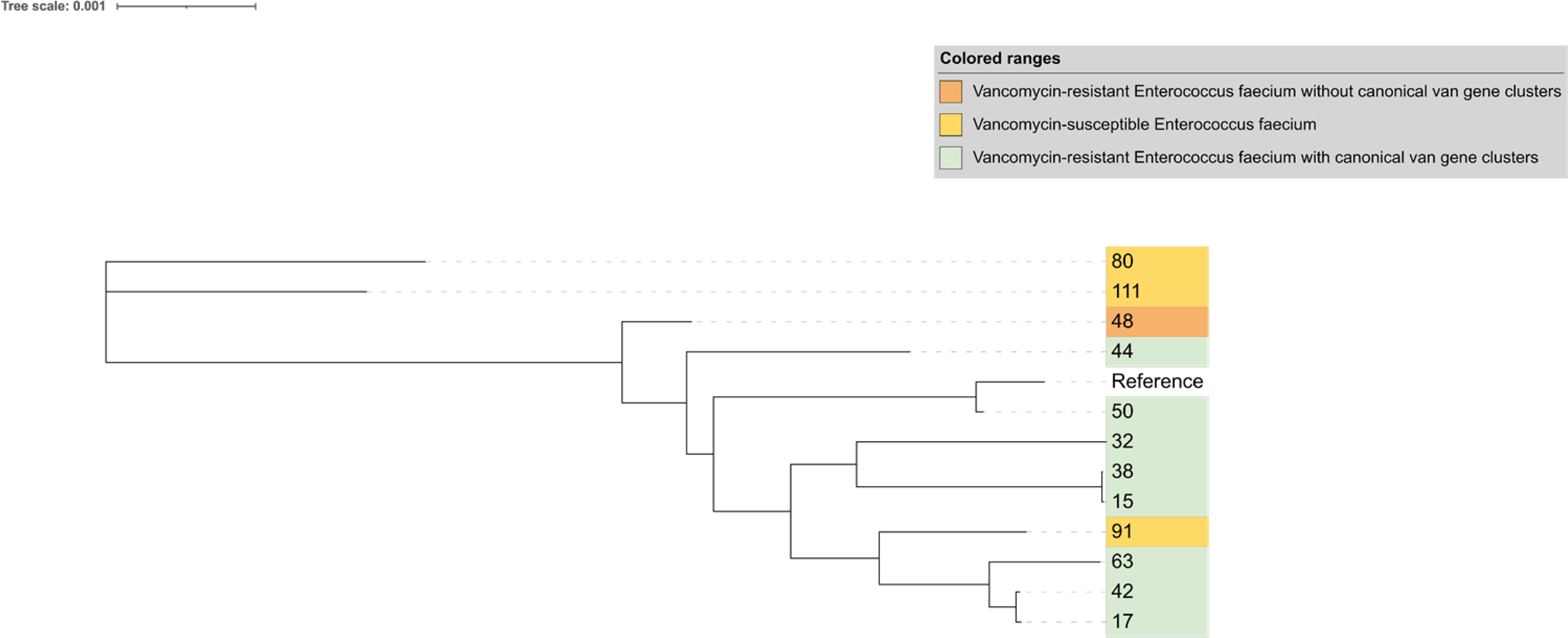
Whole-genome phylogenetic tree for Ddl gene comparator strain screening. This phylogenetic tree comprised vancomycin-resistant strain 48, three hospital-derived vancomycin-susceptible strains (111, 91, 80) and reference strains with canonical van gene clusters. The close clustering of strain 48 with the three susceptible strains supported its selection as a comparator for Ddl sequence analysis.
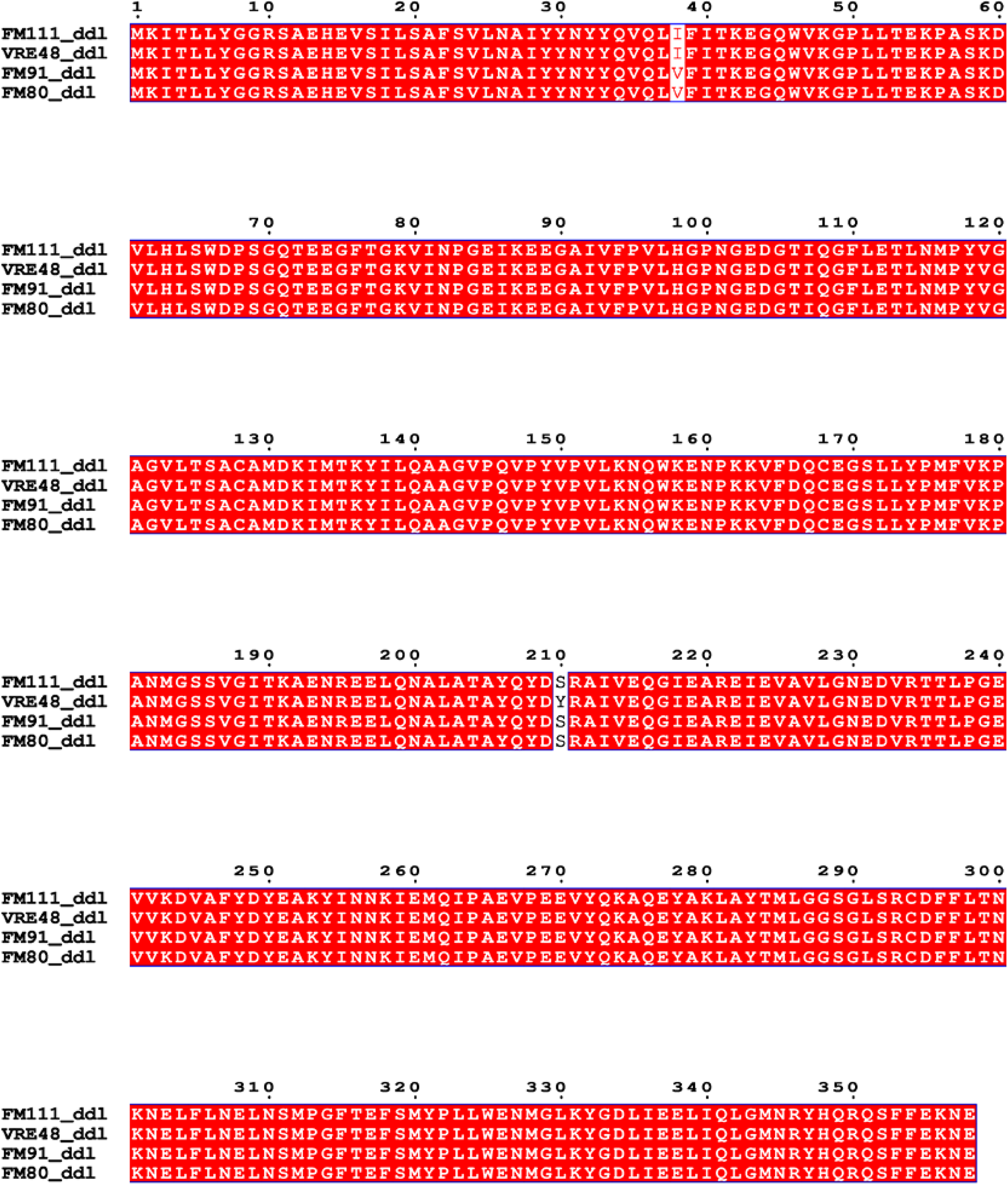
Alignment of Ddl protein sequences from Enterococcus strains. Conserved amino acid residues among strains FM111, VRE48, FM91 and FM80 (shaded in red). Numbers above indicate amino acid positions.
Discussion
This study analyzed the clinical, resistance, and genomic features of VRE in a tertiary hospital in Huizhou, providing key data to support local VRE prevention, control, and clinical management.
Clinically, all VRE isolates from a tertiary hospital in Huizhou were E. faecium, consistent with other studies and indicating its predominance in hospital settings.14,35 Especially in the case of multidrug-resistant strains, these observations highlight the need to implement targeted infection control measures to limit the spread of E. faecium, as its resistance to multiple antibiotics poses more severe clinical challenges. Midstream urine accounted for the largest proportion of VRE specimen sources (54%), This distribution is consistent with that reported in other studies. 36 The high incidence of urinary tract infections may be associated with the frequent use of urinary catheters, while bloodstream VRE infections warrant focused attention, as such infections are often accompanied by higher mortality rates and more complex clinical management challenges. Previous studies have suggested that ICU admission may be a risk factor for VRE infection. 37 In addition, the detection rate of VRE in ICU patients (57.4%) was significantly higher than that in non-ICU patients (42.6%). Moreover, ICU patients had higher proportions of underlying pulmonary and central nervous system diseases, as well as exposure to invasive procedures including urinary catheterization, gastrojejunostomy tube placement, and tracheal intubation. This confirms that multiple factors in the ICU setting, such as compromised immune function of patients, increased exposure to invasive procedures, and elevated antibiotic use intensity, 38 collectively constitute high-risk conditions for VRE infection. 39 Significant differences were found between VRE and VSE groups in vancomycin use, exposure to ≥3 antibiotic classes before detection, central nervous system disease, and urinary catheterization (p < 0.05). Among these, gender, urinary catheterization, and use of ≥3 antibiotics before pathogen detection were independent factors for VRE. The association between multiple antibiotic exposure and VRE further confirms that antibiotic selection pressure is a key driver of vancomycin resistance in enterococci. 40 No significant differences between VRE and VSE groups were observed in nasogastric or jejunostomy tube placement, tracheal intubation, or deep vein catheterization (p > 0.05), although these factors differed significantly between ICU and non-ICU patients (p < 0.05), likely due to higher invasive procedure rates in ICUs. Nevertheless, reducing antibiotic use and minimizing the use or duration of indwelling devices such as urinary and central venous catheters may help decrease VRE colonization and reduce the risk of bacteremia caused by drug-resistant pathogens. 41
Resistance testing showed that all VRE isolates were resistant to ampicillin, with over 90% resistant to penicillin, levofloxacin, and teicoplanin. In contrast, linezolid and tigecycline had very low resistance rates (0% and 1.9%, respectively), consistent with previous reports.9,42 VRE showed significant resistance differences from VSE to multiple antibiotics (ampicillin, gentamicin, levofloxacin included), which reflected the intrinsic and acquired resistance traits of VRE, 43 and accounted for the severely limited therapeutic options for VRE-infected patients. Genotypic analysis identified a high prevalence of vanA, consistent with the high vancomycin resistance rate of VRE. All isolates carried the esp gene—a key virulence factor mediating biofilm formation, adhesion, colonization and immune evasion, 44 indicating that esp might be a key virulence gene driving local infections. However, virulence genes such as gelE, cylA, and hyl were not identified in the present study. Additionally, the high prevalence of aminoglycoside resistance genes (aph, aac, ant) and the macrolide resistance gene ermB further confirmed the multidrug-resistant phenotype of VRE isolates. These findings underscore the clinical urgency of strict antibiotic stewardship to curtail the transmission risk of resistance genes. 5 This observation suggested close genetic relatedness and possible clonal expansion among these strains, providing useful information for infection prevention and control.
Whole-genome phylogenetic reconstruction delineated a distinct cluster among the study isolates. Within this cluster, pairwise core-genome SNP distances were predominantly <20 SNPs, as shown in Supplementary Table S2, supporting recent shared ancestry and suggesting possible clonal expansion rather than definitive direct transmission. However, several isolates exhibited greater within-cluster SNP divergence, indicating internal genetic substructure rather than complete homogeneity. A subset of isolates also showed high genetic similarity to Japanese and Australian reference strains, suggesting cross-regional relatedness or a common ancestral source.
Most isolates in this study clustered into a distinct, compact evolutionary clade, indicating that nosocomial VRE transmission is dominated by clonal expansion, with a small number of sporadic strains. A subset of isolates showed high genetic similarity to Japanese and Australian reference strains, suggesting cross-regional transmission or a common ancestral source. The high prevalence of CC17 aligns with global epidemic trends, 45 as this clonal complex is the major hospital-associated VRE lineage with strong resistance-acquiring ability and transmission fitness. VRE molecular epidemiology differs markedly by region: vanB-harboring VRE plus ST192 and ST17 clones dominate in Germany, 46 while ST80 and ST17 are prevalent in Thailand and Taiwan, respectively.36,47 In multiple Guangdong cities (Guangzhou, Meizhou, Zhongshan, Foshan, Qingyuan), ST80 VRE with novel vanA-carrying plasmids have caused outbreaks. Classical MLST identified ST80 as the dominant ST in this study, consistent with recent regional VRE outbreaks in Guangdong, 13 indicating nosocomial VRE transmission in this hospital may be associated with provincial clonal spread and highlighting the need for regional collaborative control. Extended MLST showed CC2 and ST106 as dominant; novel STs/CCs were only sporadic and nonwidespread, potentially representing rare local variants or imported strains that require ongoing surveillance for their evolution and distribution shifts.
In this study, VRE48 exhibited a definitive vancomycin-resistant phenotype but no detectable canonical van resistance gene clusters, in stark contrast to vancomycin-resistant strains carrying such clusters—these strains had identical Ddl gene sequences and mediated resistance via van genes. 48 These clusters reprogram peptidoglycan synthesis by replacing the terminal D-Ala-D-Ala dipeptide with D-Ala-D-Lac or D-Ala-D-Ser, thereby drastically reducing the binding affinity of vancomycin to the cell wall. 49 However, other vancomycin-resistant phenotypes independent of canonical van-mediated mechanisms have been reported, including vancomycin-dependent or revertant strains associated with mutations in host-encoded cell wall biosynthesis genes. Previous studies have shown that alterations in the host Ddl gene can affect glycopeptide susceptibility by modulating the activity of D-Ala-D-Ala ligase. In vancomycin-dependent and revertant Enterococcus faecalis strains, amino acid substitutions or insertional mutations in Ddl have been reported to impair or restore D-Ala-D-Ala synthesis, thus influencing vancomycin-related phenotypes. 50 Consistent with this concept, we identified a VRE strain lacking detectable canonical van gene clusters, which harbored a unique Ser210Tyr mutation in Ddl—a mutation absent in three closely related vancomycin-susceptible strains from the same hospital environment. Although only two amino acid differences were observed in the analyzed Ddl sequences, the presence of the unique Ser210Tyr substitution in the resistant strain highlights this position as a potential determinant associated with the observed atypical resistant phenotype. Serine and tyrosine differ significantly in side chain size and physicochemical properties, and substitution at this position may induce local conformational changes, thereby altering substrate binding or the catalytic efficiency of Ddl. Such changes could indirectly affect the availability or composition of terminal D-Ala-D-Ala, thus impinging on the interaction between vancomycin and its target. While functional validation is required to establish a causal relationship, these findings suggest that subtle alterations in host-encoded D-Ala-D-Ala ligase may lead to vancomycin resistance phenotypes independent of traditional van gene clusters. This observation broadens the range of genetic factors potentially involved in glycopeptide resistance in E. faecium and underscores the importance of considering host-associated determinants in the absence of canonical resistance mechanisms.
Limitations and Future Perspectives
This study is a single-center retrospective analysis with a limited sample size, which may not fully reflect the overall epidemiology of VRE in Huizhou. No follow-up was conducted to trace transmission routes, hindering the clarification of clonal spread extent. Future multicenter prospective studies with expanded sampling, combined with epidemiological and genomic analyses, are needed to elucidate VRE transmission dynamics. Additionally, in vitro functional experiments should verify the roles of resistance genes (the Ddl Ser210Tyr mutation) to support the development of optimized control strategies.
Conclusions
This is the first systematic report on VREfm in Huizhou. Midstream urine was the main specimen, with ICU admission and polyantibiotic use as key risk factors. VRE was dominated by CC17 (ST80), carrying vanA (94.8%) and esp (100%) as core determinants. Linezolid and tigecycline remained effective. The Ddl Ser210Tyr mutation may mediate non-classical resistance. These findings inform targeted VRE prevention/control in the hospital and Guangdong-Hong Kong-Macao Greater Bay Area, emphasizing enhanced high-risk patient surveillance, standardized antibiotic use, and strengthened infection control.
Footnotes
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.