Many oral anticancer agents are recommended to be given either at least 1 h before or 2 h after a meal, according to the prescribing information. However, the effect of dosage timing of an oral anticancer agent with reference to food intake on anticancer treatment remains unclear. As shown by the literature survey and labeling analysis for oral anticancer drugs approved by the US Food and Drug Administration from 2010 to 2016, labeling information regarding dosage timing for several anticancer drugs appeared not be optimum, leading to suboptimal bioavailability and plasma drug concentrations. This supports a call to regularly recalibrate the labeling information for dosage timing of oral anticancer medications to minimize the risks of compromised efficacy or unintended toxicities.
A majority of oral anticancer drugs are instructed to be given without regard to food intake according to the prescribing information. Even when some drugs are indicated to be taken without food, either at least 1 h before or 2 h after food is generally recommended. Nonetheless, it is largely unclear as to whether different timings of an oral anticancer agent administration in relation to food intake would lead to different efficacy or safety profiles. Recently, post-prandial dosing has been shown to provide substantially higher plasma lapatinib concentrations than pre-prandial dosing,1 raising the concern that the dosage timing with respect to food would greatly affect bioavailability and plasma concentrations of a specific anticancer agent, and subsequently, result in suboptimal treatment outcomes.
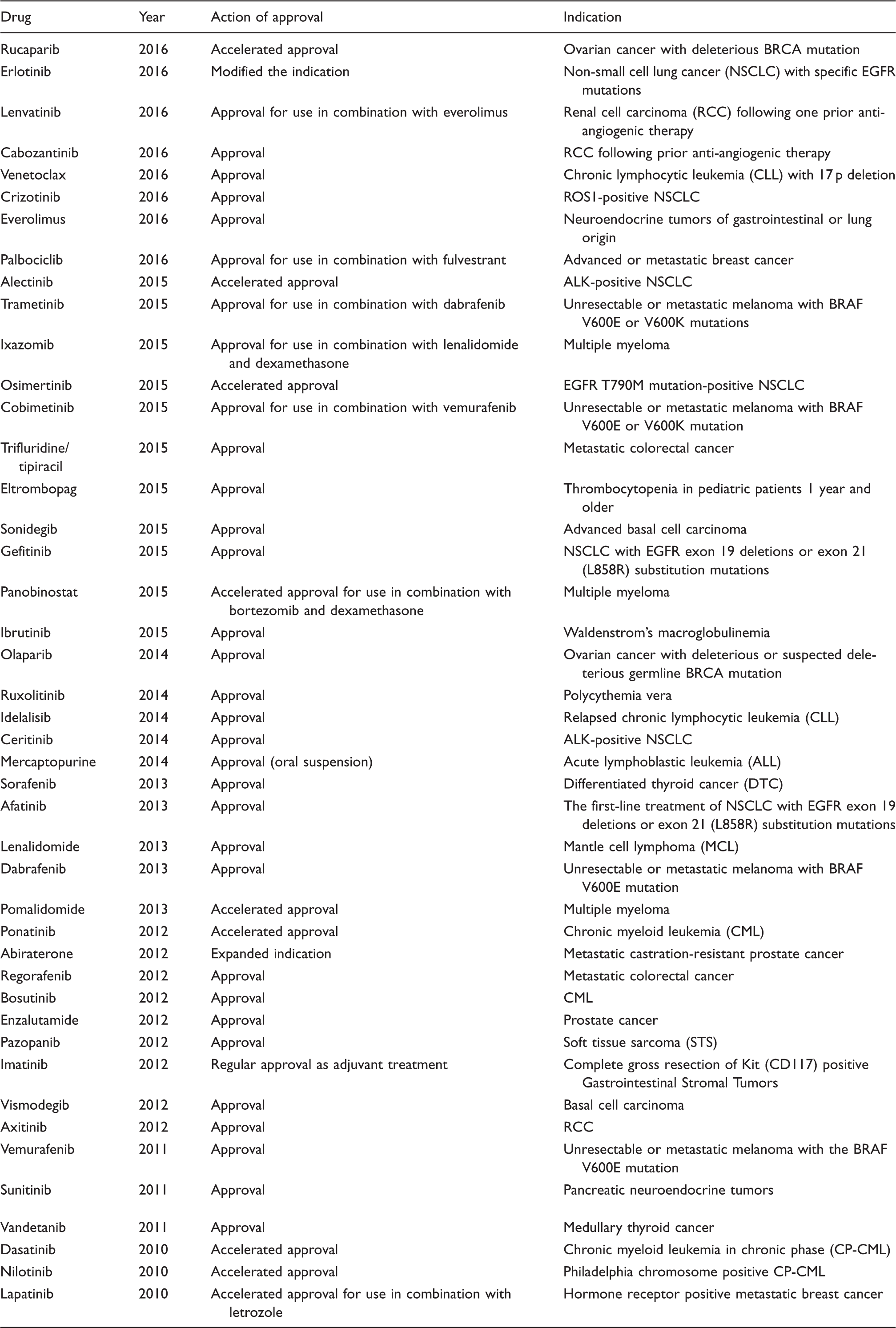
To revisit the effect of dosage timing on the bioavailability and plasma drug concentrations of a specific anticancer medication between the labeled pre-prandial and post-prandial conditions, we conducted a literature survey and labeling analysis for oral anticancer drugs approved by the US Food and Drug Administration (FDA) from January 2010 to December 2016. There were 44 oral anticancer drugs (Table 1) approved during the period according to the 140 approval records in the FDA dataset of Hematology/Oncology (Cancer) Approvals.2–6 Public available regulatory review and the latest prescribing information for these drugs were extracted from the Drug@FDA database (https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm). The literature pertaining to the dosage timing in relation to food was captured using PubMed.
Oral anticancer agents approved by the US Food and Drug Administration from 2010 to 2016.2–6
Drug
Year
Action of approval
Indication
Rucaparib
2016
Accelerated approval
Ovarian cancer with deleterious BRCA mutation
Erlotinib
2016
Modified the indication
Non-small cell lung cancer (NSCLC) with specific EGFR mutations
Lenvatinib
2016
Approval for use in combination with everolimus
Renal cell carcinoma (RCC) following one prior anti-angiogenic therapy
Cabozantinib
2016
Approval
RCC following prior anti-angiogenic therapy
Venetoclax
2016
Approval
Chronic lymphocytic leukemia (CLL) with 17 p deletion
Crizotinib
2016
Approval
ROS1-positive NSCLC
Everolimus
2016
Approval
Neuroendocrine tumors of gastrointestinal or lung origin
Palbociclib
2016
Approval for use in combination with fulvestrant
Advanced or metastatic breast cancer
Alectinib
2015
Accelerated approval
ALK-positive NSCLC
Trametinib
2015
Approval for use in combination with dabrafenib
Unresectable or metastatic melanoma with BRAF V600E or V600K mutations
Ixazomib
2015
Approval for use in combination with lenalidomide and dexamethasone
Multiple myeloma
Osimertinib
2015
Accelerated approval
EGFR T790M mutation-positive NSCLC
Cobimetinib
2015
Approval for use in combination with vemurafenib
Unresectable or metastatic melanoma with BRAF V600E or V600K mutation
Trifluridine/ tipiracil
2015
Approval
Metastatic colorectal cancer
Eltrombopag
2015
Approval
Thrombocytopenia in pediatric patients 1 year and older
Sonidegib
2015
Approval
Advanced basal cell carcinoma
Gefitinib
2015
Approval
NSCLC with EGFR exon 19 deletions or exon 21 (L858R) substitution mutations
Panobinostat
2015
Accelerated approval for use in combination with bortezomib and dexamethasone
Multiple myeloma
Ibrutinib
2015
Approval
Waldenstrom’s macroglobulinemia
Olaparib
2014
Approval
Ovarian cancer with deleterious or suspected deleterious germline BRCA mutation
Ruxolitinib
2014
Approval
Polycythemia vera
Idelalisib
2014
Approval
Relapsed chronic lymphocytic leukemia (CLL)
Ceritinib
2014
Approval
ALK-positive NSCLC
Mercaptopurine
2014
Approval (oral suspension)
Acute lymphoblastic leukemia (ALL)
Sorafenib
2013
Approval
Differentiated thyroid cancer (DTC)
Afatinib
2013
Approval
The first-line treatment of NSCLC with EGFR exon 19 deletions or exon 21 (L858R) substitution mutations
Lenalidomide
2013
Approval
Mantle cell lymphoma (MCL)
Dabrafenib
2013
Approval
Unresectable or metastatic melanoma with BRAF V600E mutation
Pomalidomide
2013
Accelerated approval
Multiple myeloma
Ponatinib
2012
Accelerated approval
Chronic myeloid leukemia (CML)
Abiraterone
2012
Expanded indication
Metastatic castration-resistant prostate cancer
Regorafenib
2012
Approval
Metastatic colorectal cancer
Bosutinib
2012
Approval
CML
Enzalutamide
2012
Approval
Prostate cancer
Pazopanib
2012
Approval
Soft tissue sarcoma (STS)
Imatinib
2012
Regular approval as adjuvant treatment
Complete gross resection of Kit (CD117) positive Gastrointestinal Stromal Tumors
Vismodegib
2012
Approval
Basal cell carcinoma
Axitinib
2012
Approval
RCC
Vemurafenib
2011
Approval
Unresectable or metastatic melanoma with the BRAF V600E mutation
Sunitinib
2011
Approval
Pancreatic neuroendocrine tumors
Vandetanib
2011
Approval
Medullary thyroid cancer
Dasatinib
2010
Accelerated approval
Chronic myeloid leukemia in chronic phase (CP-CML)
Nilotinib
2010
Accelerated approval
Philadelphia chromosome positive CP-CML
Lapatinib
2010
Accelerated approval for use in combination with letrozole
Hormone receptor positive metastatic breast cancer
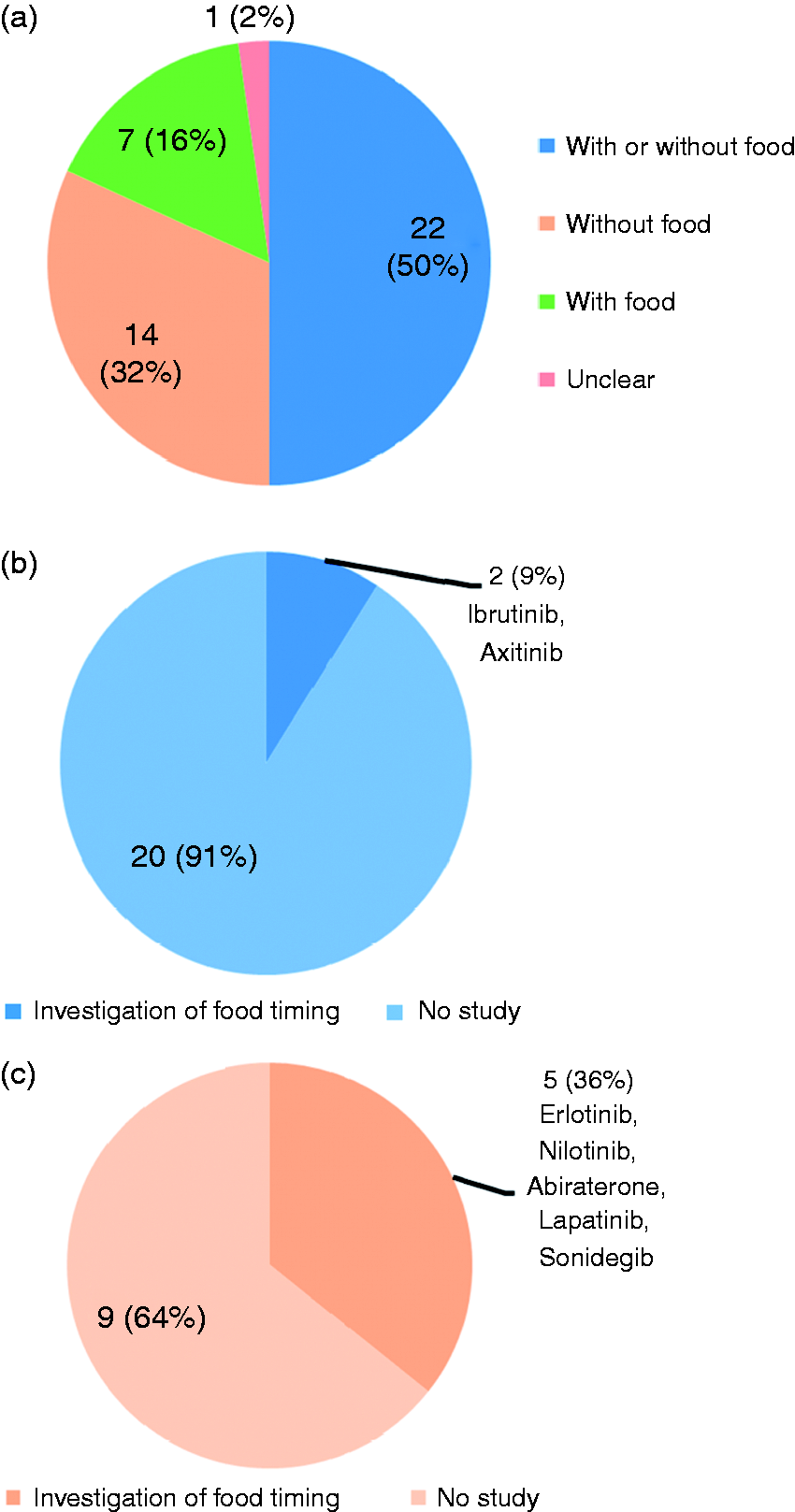
As shown in Figure 1, 22 (50%) of the 44 oral oncology medications were approved to be administrated without restrictions on food intake. The effect of dosage timing in terms of pre-prandial versus post-prandial dosing on bioavailability was investigated in only 2 (9%) drugs, i.e. ibrutinib7 and axitinib,8 among them. For 14 (32%) drugs who were indicated to be given without food, studies on food timing were performed in only 5 (36%) of those, i.e. erlotinib,9 abiraterone,10 nilotinib,11 lapatinib,12 and sonidegib.13 Comparative data of bioavailability in terms of peak plasma concentration and systemic drug exposure (i.e. area under the plasma concentration-time curve, AUC) under the recommended pre-prandial and post-prandial conditions from the literature are summarized in Table 1.
Types of food recommendation for oral anticancer agents. (a) Proportion of each type of food recommendation. (b) Investigation of dosage timing of drugs in relation to food among those labeled to be given without regard to food. (c) Investigation of dosage timing of drugs among those labeled to be given without food.
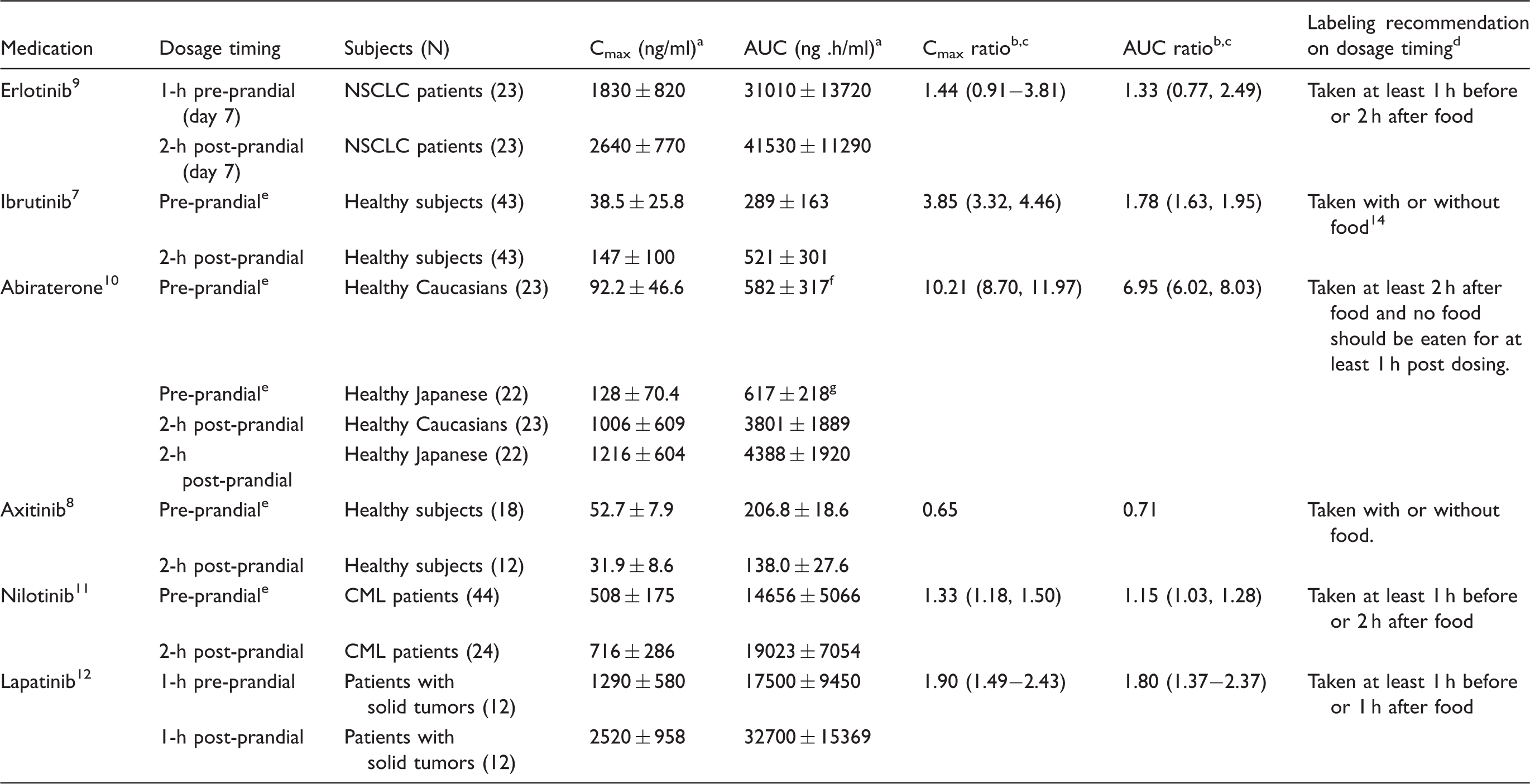
For erlotinib9 and nilotinib,11 peak plasma concentrations were increased by 33% and 44%, respectively, when administered 2 h after food compared with those administered at least 1 h before food. Likewise, systemic drug exposure (i.e., area under the plasma concentration-time curve, AUC) for the two drugs were slightly elevated by 15%–33% under 2-h post-prandial state than those under 1-h pre-prandial fasting state. For axitinib,8 either peak plasma concentration or systemic drug exposure were slightly decreased by 35% or 29%, respectively, when administered 2 h after food relative to those administered at least 1 h before food. Similarly, on the basis of a population pharmacokinetic analysis,13 the dosage timing in relation to food (i.e. 2-h post-prandial dosing versus at least 1-h pre-prandial dosing) did not have a statistically significant impact on sonidegib bioavailability. Accordingly, the differences in bioavailability of erlotinib, nilotinib, axitinib, and sonidegib between the recommended pre-prandial and post-prandial conditions appeared not to be of clinical relevance.
However, the bioavailability of lapatinib was substantially increased by 80%– 90% when taken 1 h before a low-fat meal compared to that under 1-h post-prandial conditions.12 Moreover, for ibrutinib7 and abiraterone,10 4 to 10 times increases in peak plasma drug concentrations and 2 to 7 times increases in systemic drug exposure were observed when administered 2-h post a meal than that administrated at least 1 h prior to a meal. Similar to lapatinib, substantial differences in plasma concentrations of abiraterone and ibrutinib between pre-prandial and post-prandial dosing were seen even when they were given according to the labeling instruction for drug administration (Table 2). The impaired bioavailability of lapatinib, ibrutinib, and abiraterone, under the labeled post-prandial conditions might lead to compromised efficacy and even failure of anticancer treatment in some individual patients.
Effect of dosage timing on peak concentration and systemic exposure of oral anticancer agents.
AUC: area under the plasma concentration-time curve; CI: confidence interval; Cmax: peak drug concentration in plasma; CML: chronic myeloid leukemia; NSCLC: non-small cell lung cancer.
Optimal instructions for dosage timing in relation to food could improve the balance of benefits against risks of anticancer treatment and patient compliance, which are crucial for oncological clinical practice. Unfortunately, as shown by the literature survey and labeling analysis, labeling information regarding dosage timing for several anticancer drugs, such as lapatinib, ibrutinib, and abiraterone, may not be optimum, leading to suboptimal bioavailability and plasma drug concentrations. This supports a call to regularly recalibrate the labeling information for dosage timing of oral anticancer medications in order to minimize the risks of compromised efficacy or unintended toxicities associated with suboptimal plasma drug concentrations. Also, when a remarkable food effect is observed, additional food–drug interaction studies evaluating the impact of drugs given at different time points before and after a meal should be conducted to ensure optimum dosing labeling.15,16 Furthermore, oncologists should be aware of the potential suboptimal clinical outcome when different dosage timings with reference to food are implemented.
Footnotes
Disclosure of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by the National Natural Science Foundation of China (No. 81603209).
References
1.
OhgamiMBandoHIshiguroHet al.Effect of dose timing on the blood concentration of lapatinib in patients with breast cancer. Ann Oncol2017; 28: 2888–2889.
de JongJSukbuntherngJSkeeDet al.The effect of food on the pharmacokinetics of oral ibrutinib in healthy participants and patients with chronic lymphocytic leukemia. Cancer Chemother Pharmacol2015; 75: 907–916.
8.
PithavalaYKChenYTohMet al.Evaluation of the effect of food on the pharmacokinetics of axitinib in healthy volunteers. Cancer Chemother Pharmacol2012; 70: 103–112.
9.
KatsuyaYFujiwaraYSunamiKet al.Comparison of the pharmacokinetics of erlotinib administered in complete fasting and 2 h after a meal in patients with lung cancer. Cancer Chemother Pharmacol2015; 76: 125–132.
10.
InoueKShishidoAVaccaroNet al.Pharmacokinetics of abiraterone in healthy Japanese men: dose-proportionality and effect of food timing. Cancer Chemother Pharmacol2015; 75: 49–58.
11.
TanakaCYinOQSethuramanVet al.Clinical pharmacokinetics of the BCR-ABL tyrosine kinase inhibitor nilotinib. Clin Pharmacol Ther2010; 87: 197–203.
12.
DevrieseLAKochKMMergui-RoelvinkMet al.Effects of low-fat and high-fat meals on steady-state pharmacokinetics of lapatinib in patients with advanced solid tumours. Invest New Drugs2014; 32: 481–488.
13.
GoelVHurhESteinAet al.Population pharmacokinetics of sonidegib (LDE225), an oral inhibitor of hedgehog pathway signaling, in healthy subjects and in patients with advanced solid tumors. Cancer Chemother Pharmacol2016; 77: 745–755.