
Abstract
The Food and Drug Administration has recently approved two autologous chimeric antigen receptor T-cell immunotherapies tisagenlecleucel (Kymriah™) and axicabtagene ciloleucel (Yescarta™) for patients with advanced lymphocytic malignancies. Both immunotherapies target the CD19-positive B-cell neoplasms. Kymriah™ is indicated for the treatment of relapsed or refractory acute lymphoblastic leukemia and large B-cell lymphoma. Yescarta™ is indicated for lymphoma only. Although the new therapy offers a promise for patients with advanced disease, it is associated with adverse events including neurotoxicity and cytokine release syndrome, which can be fatal and may require a high level of multidisciplinary supportive care. Due to the risks, both Kymriah™ and Yescarta™ are subject to Risk Evaluation and Mitigation Strategy (REMS) protocols. As active members of multidisciplinary clinical teams, pharmacists are likely to be responsible for the execution of Kymriah™ and Yescarta™ REMS programs. This manuscript describes foundational science and clinical knowledge of chimeric antigen receptor T-cell immunotherapies, common therapy-specific toxicities and REMS requirements for Kymriah™ and Yescarta™ in relation to practice of pharmacy.
Keywords
Introduction
With the recent Food and Drug Administration (FDA) approval of two novel immunotherapies, there is a promise for patients with advanced lymphocytic malignancies. Both novel immunotherapies target B-cell neoplasms that express CD19 proteins on cell surfaces, and are approved for use after two or more lines of systemic chemotherapy.1,2 Tisagenlecleucel (Kymriah™; Novartis Pharmaceuticals) is indicated for the treatment of relapsed or refractory (r/r) B-cell precursor acute lymphoblastic leukemia (ALL) in patients who are 25 years of age or younger (approved, August 2017) and large B-cell lymphoma in adults (approved, May 2018). 1 Axicabtagene ciloleucel (Yescarta™; Kite Pharmaceuticals, October 2017) bears an indication for r/r large B-cell lymphoma in adults only. 2 Both immunotherapies demonstrated induction of remission in a substantial number of patients and were observed in the blood of patients who responded to the treatment for more than a year.1–4 This breakthrough outcome was the result of years of research in molecular pharmacology that yielded a novel system of infusion immunotherapy—adoptive transfer of chimeric antigen receptor (CAR) T-lymphocytes. 5 However, most patients receiving a CAR T-cell infusion will develop therapy-triggered adverse events that, in some cases, have been severe requiring a high level of multidisciplinary supportive care 6 and warranting Black Box Warnings for both products.1,2
To ensure that the benefits of novel immunotherapies outweigh the risk of toxicities, FDA instituted Risk Evaluation and Mitigation Strategy (REMS) program for both tisagenlecleucel 7 and axicabtagene ciloleucel. 8 Both REMS programs require institution’s certification prior to dispensing of therapy. A crucial component of REMS attestation is designation of an authorized representative responsible for all processes and procedures associated with the immunotherapies, and availability of tocilizumab (Actemra®; Genentech Inc., August 2017) used for reversal of life-threatening syndromes. Pharmacists being active members of multidisciplinary clinical teams are often the key personnel involved in execution of REMS programs.
This article describes foundational science, clinical knowledge, common therapy-specific toxicities, and REMS requirements associated with tisagenlecleucel (Kymriah™) and axicabtagene ciloleucel (Yescarta™) as they relate to practice of pharmacy.
Foundational knowledge of tumor immunity
To understand the concept of CAR T-cell therapy, it is important to recognize tumor immunity and how it occurs naturally. One of the critical functions of the immune system is to identify and eradicate clones of transformed cells before they become tumors and to destroy tumors after they are formed. This process is known as immune surveillance. 9 Immune cells that are responsible for killing tumors include natural killer cells and T cells.
To mount anti-tumor response, the immune system must be able to recognize tumors, which will then be destroyed by effector cells such as cytotoxic T lymphocytes (CTLs). Successful generation of tumor-specific CTLs is therefore critical and is relying on optimal activation of naïve T-cells by professional antigen presenting cells (APCs) such as dendritic cells (DCs). In brief, host immature DCs engulf tumor proteins and migrate to draining lymph nodes. The engulfed tumor proteins are processed and presented on membrane surface of the DCs in the form of human leukocyte antigen: tumor-associated antigen (HLA: TAA) complex. In the lymphoid organs, the DCs present tumor antigens to CD8+ and CD4+ naive T-cells and activate them through two critical signals known as signal 1 and signal 2 (Figure 1). By receiving both signals, the naïve T-cells become activated and then differentiated into effector cells i.e. tumor-specific CTLs and T-helper cells, respectively. These effector cells, CTLs in particular, then migrate back to tumor sites and kill tumor cells by releasing cytotoxic granules containing granzymes and other cytotoxic proteins (Figure 1). It is important to note that T-helper cells, specifically the Th1 cells, are needed for (1) optimal and sustained CTL responses and (2) induction and maintenance of CD8+ memory. Hence, both CD8+ and CD4+ T-cells are vital in induction and maintenance of tumor immunity.
Immune response to tumor. Immature DCs uptake tumor proteins and migrate to draining lymph nodes where they become mature. The mature DCs interact and engage with naïve T-cell in the lymph nodes through signal 1 (HLA: TAA complex–TCR interaction) and signal 2 (co-stimulatory molecule–CD28 interaction) to activate the naïve T-cells. The generation of signal 1 alone results in inactivation of the T-cells. Activated T-cells become effector cells—CTLs and T-helper cells (not shown). The effector CTLs leave the lymph nodes, migrate back to tumors, and kill tumor cells.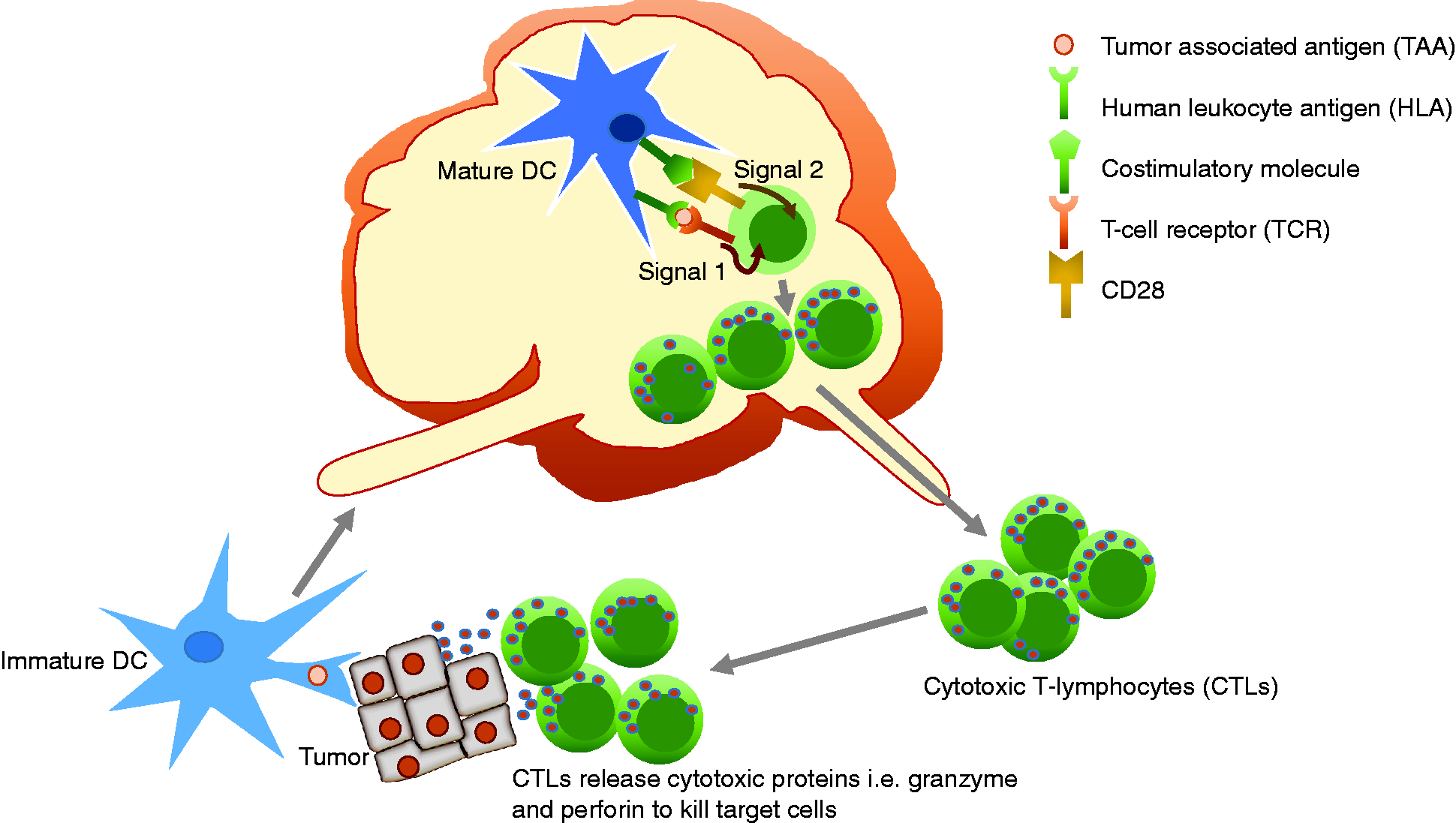
Mature DCs express cell surface molecules necessary for activation of naïve CD8+ T-cells. These surface molecules include class I HLA complexed with a peptide derived from tumor antigens, and costimulatory molecules such as B7-1 (CD80), B7-2 (CD86), and CD40. To date, additional costimulatory molecules have been identified, which include 4-1BB (CD137) and OX40 (CD134). Class I HLA: TAA complex on the surface of DC interacts with T-cell receptor (TCR), providing antigen recognition and T-cell activation signal known as “signal 1”. Costimulatory molecules i.e. CD80 and CD86 on DC surface also interact with CD28 on the membrane surface of naïve T-cells providing additional T-cell activating signal, known as “signal 2”. As a result, these naïve CD8+ T-cells are activated and differentiated into effector CTLs (Figure 1).
Pharmacology of chimeric antigen receptor T-cells
Adoptive cell transfer therapy (ACT) involves administration of immune cells with direct anticancer activity to the cancer-bearing patient (host).
5
One type of ACT is CAR T-cell therapy. Prior to CAR T-cell therapy era, a general approach for adoptive transfer of TAA-specific T cells was done by isolating TAA-specific T-cells from a cancer patient and expanding them ex vivo before infusing them back into the patient.
10
This approach was limited by a necessity to isolate tumor reactive T cells from each individual patient.
11
Additionally, these TAA-specific T cells are anergic, and difficult to expand ex vivo because they require the relevant HLA allele to be recognized by TCRs of the TAA-specific T cells
12
(Figure 2). More importantly, the TCRs of these TAA-specific T-cells have substantially lower antigen affinity vs. TCRs directed against viral-specific antigens.
13
To overcome these challenges, genetically modified CAR expressing T-cells have been developed. These T-cells undergo bioengineering process to express single-chain antibody variable fragment (scFv) designed to recognize tumor-associated antigenic epitope with high affinity in a chimeric T-cell receptor construct described below. Because the scFv is designed to recognize and bind to the specific TAA directly, the CAR T-cells bypass the HLA restriction (HLA-restricted antigen recognition) therefore effective in tumors avoiding CTL recognition via reduced expression of HLA molecules.
Left, T-cell receptor (TCR)–CD3 complex. Most T-cells express α:β chains on membrane surface as their TCRs. These TCRs are associated with CD3 complex that contains one CD3γ chain, one CD3δ chain, and two CD3ɛ chains, and two CD3ζ chains. Right, interaction between antigen presenting cell such as dendritic cell and CD8 T-lymphocyte. Stimulation of TCR is triggered by HLA:peptide (e.g. TAA) complex. Engagement of the TCR with class I HLA:TAA complex initiates signaling cascades that lead to ZAP-70 activation, resulting in T-cell activation, proliferation, differentiation, and cytokine production.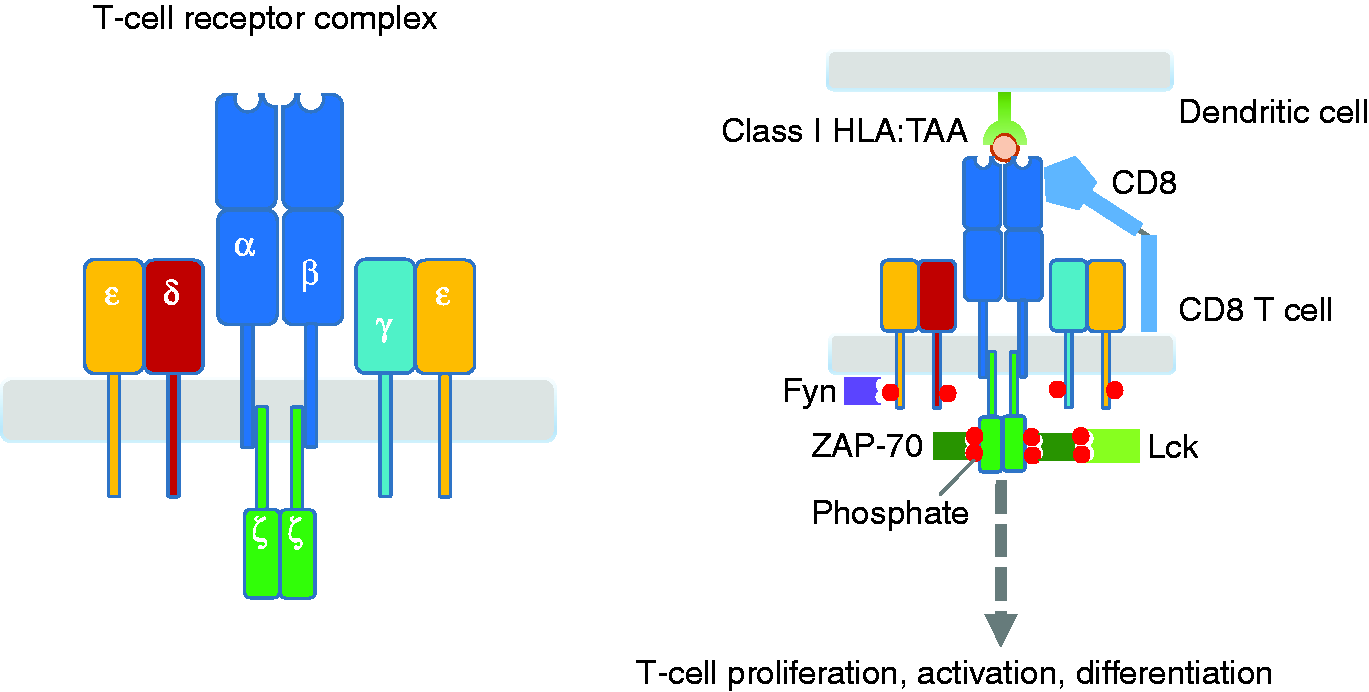
As aforementioned, activation of T-cells requires the relevant HLA allele that is recognized by TAA-specific TCR. This canonical TCR consists of α:β heterodimer that is associated with the CD3 complex (CD3γ, CD3δ, and two CD3ɛ) and CD3 zeta (ζ) chains (Figure 2, left). When TCRs become clustered upon binding the HLA:peptide complex, a receptor-associated kinase, Fyn, is activated, leading to phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) of the CD3 complex, and those on the CD3ζ chain. These events allow the zeta-chain-associated protein kinase 70 (ZAP-70) to bind to phosphorylated ITAM of the CD3ζ chain. Once the co-receptor (i.e. CD4 and CD8) binds the HLA:peptide complex, which in turn, brings another receptor-associated kinase, Lck into the complex, Lck then phosphorylates and activates the ZAP-70. Consequently, T cells are activated, proliferate, and differentiated into the effector T cells (Figure 2, right).
In contrast to the canonical TCR, CAR is a genetically engineered single polypeptide chain receptor. It consists of extracellular domain (scFv), which recognizes TAA, spacer region, transmembrane (TM) domain, and intracellular domains derived from molecules necessary for T-cell signaling. The scFv is an antigen binding site, which consists of variable regions of the heavy (VH) and light chains (VL) of immunoglobulins that are linked together through a short peptide segment.14,15 The intracellular domains contain CD3ζ chain signaling domain (providing signal 1) and the signaling motifs present in the cytoplasmic domain of the costimulatory molecules such as 4-1BB, OX-40 and CD28 (providing signal 2). These intracellular domains are responsible for T-cell activation. Different compositions of CAR intracellular domains have led to creation of three generations of CAR constructs (Figure 3).
16
The first-generation CARs have two major limitations—limited T-cell proliferation and diminished cytolytic activities
17
due to the lack of T-cell co-stimulation.
18
These limitations have led to the development of the second- and subsequently the third-generation CARs in an attempt to improve the efficacy and effectiveness of CAR T-cell therapy.
Three generations of chimeric antigen receptors (CARs). First-generation CARs are composed of extracellular single-chain antibody variable fragment (scFv) that are specific to TAA, transmembrane domain (TM) and cytoplasmic signaling domain CD3ζ. Second-generation CARs incorporate a costimulatory domain i.e. CD28 or 4-1BB into the cytoplasmic tail in addition to CD3ζ. Third-generation CARs incorporate two costimulatory domains such as CD28 and 4-1BB or CD28 and OX40.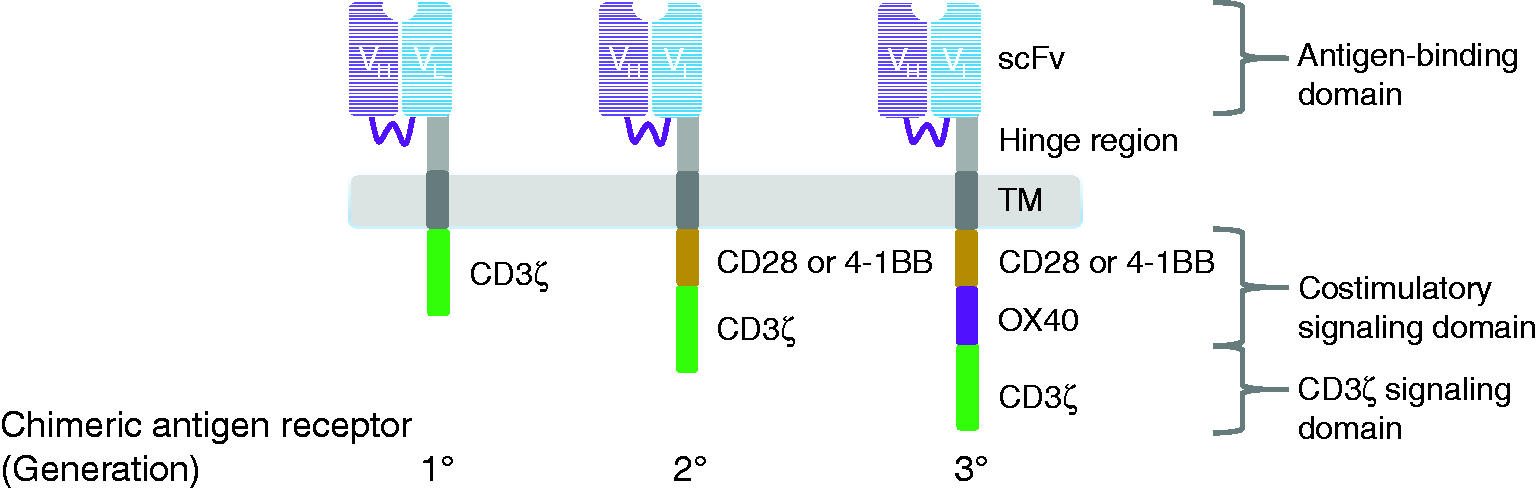
First generation CAR: The intracellular domain of the first-generation CAR contains only CD3ζ chain. Several clinical trials employing the first generation of CAR T-cells have demonstrated limited success in various types of cancer including renal cell carcinoma, non-Hodgkin’s lymphoma, and ovarian cancer. The challenge of this generation is modest antitumor activity and difficulty in expanding the CAR expressing T-cells. 12
Second generation CAR: To overcome the issues of the first generation, an additional costimulatory molecule i.e. CD28 or 4-1BB is incorporated into the signaling domain of CARs. This enhancement markedly increases production of IL-2, which in turn, stimulates proliferation and improves effector functions of CAR T-cells. 19
The two recently approved immunotherapies belong to the second generation of CAR. The two products differ by costimulatory molecules in their cytoplasmic tails; Yescarta’s tail contains CD28 whereas Kymriah’s – 4-1BB.1,2 Currently, it is unknown which costimulatory molecule is more beneficial. Data indicate that CD28-based CARs exhibit rapid expansion and boost effector functions, whereas constructs with 4-1BB generate greater persistence and longevity. 20
Third generation CAR: In the third generation, two signaling domains of costimulatory molecules are added into the CAR construct. The costimulatory molecules in the third generation CARs that have been used thus far are a combination of CD28 and 4-1BB or CD28 and OX40. In theory, a combination of two costimulatory molecules might further enhance the effectiveness of CAR T-cell therapy. 21
Pharmacokinetics/cellular kinetics of CAR T-cells
The units of pharmacokinetic parameters for CAR T-cells appear to be different from that for conventional drugs. For example, unit of the maximum plasma concentration (Cmax) may be expressed as either copies per microgram (copies/mcg) or cells/microliter (cells/µL) instead of usual expression of drug concentration in mg/mL.
Following axicabtagene ciloleucel infusion, the Cmax were 43.6 cells/µL and 21.2 cells/µL in complete remission and non-responding patients, respectively. The anti-CD19 CAR T-cells peaked within 1–2 weeks followed by a decline to near baseline levels by three months. The AUC0-28d were 557.1 days*cells/µL vs. 222.0 days*cells/µL, in complete remission and non-responding patients, respectively. 2
After administration of tisagenlecleucel, the tisagenlecleucel transgene was evaluated as copies per microgram (copies/mcg). In pediatric and young adult patients with r/r ALL, the Cmax of CAR transgene were 34,700 copies/mcg and 20,000 copies/mcg in patients with complete remission and non-respondents, respectively. The median time to maximum expansion (Tmax) was approximately 10 and 20 days in complete remission and non-responding patients, respectively while the AUC0-28d were 318,000 copies/mcg*day in complete remission patients vs. 156,000 copies/mcg*day in nonresponding patients. Tisagenlecleucel was also detected in bone marrow and was measurable beyond two years. 1
Clinical use of CAR T-cell immunotherapies and issues in supportive care
Both axicabtagene ciloleucel (Yescarta™) and tisagenlecleucel (Kymriah™) are prepared and administered through five steps, which might take up to four weeks to complete. In general, the steps are similar: (1) harvesting of patient’s T-cells via standard leukapheresis; (2) T-cells activation; (3) T-cells transduction; (4) T-cells expansion; (5) CAR T-cell infusion back into the patient. 22 The manufacturing differs at steps 2 and 3.1,2 For axicabtagene ciloleucel, T-cells are activated with anti-CD3 antibody in the presence of IL-2 and then transduced with the replication incompetent retroviral vector that contains the anti-CD19 CAR transgene. With tisagenlecleucel, T-cells are transduced with lentiviral vector first, and then activated with anti-CD3/CD28 antibody-coated beads that mimic DC signals (Figure 1). At step 4, the transduced T-cells are expanded in cell culture, washed, formulated into a suspension, and cryopreserved. The cells are tested for sterility and released for shipping as a frozen suspension in a patient-specific bag. A few days prior to a single dose intravenous CAR T-cell transfusion, most patients are pre-treated with a lymphodepleting cytotoxic regimen. The regimen usually consists of cyclophosphamide 500 mg/m2 IV and fludarabine 30 mg/m2 IV. The transfusion can occur in in-patient setting or in an outpatient infusion clinic, contingent upon institution’s experience with the immunotherapies.3,23 REMS Yescarta™ and Kymriah™ allow patient enrollment and dispensing of the immunotherapies only at the authorized treatment centers, which are hospitals and affiliated clinics that have registered with REMS and fulfilled REMS requirements.7,8
Once the autologous CAR T-cells are infused and start binding to their specific target, a conformational change of CAR is induced, ultimately leading to activation of CAR T-cells. The activated CAR T-cells (effector cells) release perforin and granzymes that attack and eradicate tumor cells.
The CAR T-cell immunotherapies have shown substantial clinical efficacy in adult and pediatric patients with r/r hematologic malignancies previously treated with two or more lines of systemic therapy. Administration of axicabtagene ciloleucel to adult patients with r/r aggressive B-cell non-Hodgkin’s lymphoma has resulted in overall response rate (ORR) of 82%, with complete response (CR) of 54%, and a 6-month progression-free survival (PFS) of 49%. Median time to response was 1 month (range 0.8–6 months). 24 In adult patients with r/r diffuse large B-cell lymphoma (DLBCL) who received tisagenlecleucel, the ORR was 53.1% with CR of 39.5% and PR of 13.6%. 25 In a clinical trial evaluating the overall remission rate in young patients with r/r B-cell ALL treated with tisagenlecleucel, complete remission was observed in 60% of participants, and 21% of treated patients had complete remission with incomplete hematologic recovery. Ninety-five percent of patients with complete remission with or without complete hematologic recovery were negative for minimal residual disease (MRD) by day 28. 26
The activation of T-cells mediated by CARs is associated with the massive release of proinflammatory cytokines including IL-1, IL-6, IL-8, IL-15, and IFNγ, which manifests as a clinical phenomenon of cytokine-release syndrome (CRS). 6 CRS is diagnosed based on a presentation with fever, malaise, anorexia, and/or myalgias that can progress to capillary leak, hypoxia with pulmonary edema, neutropenic sepsis, hepatic, and/or renal dysfunction over the course of several days. 27 The CRS poses a high risk of progressing to a fatal outcome and might represent medical emergency. 28 In most instances, CRS has occurred within 1–14 days after tisagenlecleucel infusion 27 and within the first week in patients treated with axicabtagene ciloleucel. 24 The median time to syndrome resolution was eight days for both therapies.24,29
The any grade syndrome was reported in over 90% of patients on axicabtagene ciloleucel, with 13% developing grade 3 or higher.2,24 Seventy-nine percent of patients with r/r ALL and 74% of patients with r/r DLBCL receiving tisagenlecleucel developed any grade CRS, including grade 3 or higher in 49% of patients with r/r ALL and in 23% of patients with r/r DLBCL. 1 Severity of CRS is driven by high disease burden, patients’ demographics, and comorbidities. It might be less severe in younger individuals than in adults. 23
Existing evidence suggests that IL-6 plays a pivotal role of in the pathogenesis of CRS.26,29–32 IL-6 has pleiotropic effects including immune response, inflammation, and hematopoiesis. 33 This cytokine induces production of acute-phase proteins such as C-reactive protein (CRP), fibrinogen, and hepcidin, whereas it reduces the biosynthesis of albumin and cytochrome P450 (CYP450) enzymes in the liver. 34 Due to IL-6 critical role in the pathogenesis of CRS, recombinant humanized anti-human IL-6 receptor IgG1κ monoclonal antibody – tocilizumab (Actemra®, Genentech, Inc., August 2017) was incorporated in the protocols for clinical trials and later approved for patients with CAR T-cell-induced severe or life-threatening CRS.28,35 A retrospective analysis of pooled data from the clinical trials demonstrated the response rate to tocilizumab between 53% and 69% with fever and tachycardia resolving rapidly, and recovery from organ dysfunction taking longer time. 28 The median time to response was about four days. No adverse reactions were attributed to administration of tocilizumab. The labeling for Actemra® advises against administration of the drug to patients with absolute neutrophil count (ANC) less than 2000/mm3 or platelets less than 100,000/mm3 or liver function tests (LFTs) above 1.5 times the upper limit of normal (ULN). It suggests weighing the benefit of treating the CRS against the risk of short-term exposure to tocilizumab since the mechanism of tocilizumab-induced neutropenia is unlikely to be related to myelosuppression.28,35
Another common type of CAR T-cell-related toxicities is neurological complications, which are often referred to as CAR T-cell-related encephalopathy syndrome (CRES). 24 All grade neurotoxicities occurred in 72% of r/r ALL patients and in 58% of r/r DLBCL patients post-therapy with tisagenlecleucel, with reactions of grade 3 or higher occurring in 21% of patients with r/r ALL and in 18% of patients with r/r DLBCL. 1 Similarly, neurotoxicities were reported in 87% of patients treated with axicabtagene ciloleucel. 2 Encephalopathy of any grade was the most commonly experienced neurotoxicity, 57% of patients, and it remained the most frequent grade 3 or higher neurological complication, 29%. 2 In patients treated with tisagenlecleucel, the incidence of serious encephalopathy was about 10% in both ALL and DLBCL cohorts while any grade encephalopathy was 34% for ALL and 16% for DLBCL patients. 1
The onset of neurological toxicity was similar for both immunotherapies, within eight weeks of infusion.1,2 Neurotoxicities can develop concurrently with CRS or without it.1,2 Unlike for CRS, the pathophysiology of this complication remains to be elucidated. It is possible that neurotoxicity is mediated by proinflammatory cytokines such as IL-1 and IL-6 that accumulate in the brain either by passive diffusion from the periphery, or by infiltration of CAR T-cells into the CNS. The toxic encephalopathy can manifest as early and delayed phases. 6 The early phase, which usually occurs within the first week of CAR T-cell infusion, might manifest as decreased attention, language difficulties and impaired handwriting. The late phase might develop in third-fourth week post-infusion as seizures, mental obtundation and increased intracranial pressure. The delayed phase is often unresponsive to tocilizumab.
Initial symptomatic support is provided with antipyretics, acetaminophen being the first choice, antibiotics per local sensitivities for neutropenic patients, fluids, vasopressors, and oxygen.1,2,6 It is recommended to place contingency orders before the initiation of immunotherapy to provide timely interventions once needed. 6
Guidance for toxicity grading, initiation of tocilizumab, and dosing of corticosteroids in the management of CRS and CRES vary between the immunotherapies since information was derived from research protocols for each respective product. 35 The FDA-approved protocol for Yescarta™ utilizes the CRS grading system proposed by Lee et al. 36 where tocilizumab is initiated for Grade 2 CRS, described as “symptoms require and respond to moderate intervention” such as hypotension responding to fluids and a low-dose pressor or oxygen requirement < 40% FiO2. 2 For patients with CRS post-Kymriah™ infusion, tocilizumab is started when “moderate to aggressive intervention” is required that is defined as rapid clinical deterioration, hemodynamic instability despite intravenous fluids and vasopressor support. 1 The threshold for initiation of corticosteroids also varies between the products. The Yescarta™ CRS grading and management guidance suggests intravenous (IV) methylprednisolone 1 mg/kg twice daily or an equivalent dose of dexamethasone as 10 mg IV every 6 h for CRS grade 2 with or without CRES if no clinical improvement in 24 h after tocilizumab. The dose of methylprednisolone is increased to 1000 mg/day for grade 4 of CRS with or without CRES, and for the patients with grade 4 of CRES methylprednisolone is administered with the first dose of tocilizumab. 2 The labeling for Kymriah™ does not recommend an increase in dosing of steroids for severe cases of CRS and does not address the specifics of CRES management. 1 In communication by Teachey et al., 23 authors express a concern that high doses of corticosteroids could reduce the therapeutic efficacy of tisagenlecleucel and refer to their positive experience with the lower doses of methylprednisolone. They advise against empirical corticosteroids for low-grade neurotoxicity and advocate tailoring treatment decisions to the type of neurotoxicity and specifics of the received CAR T-cell therapy.
Implications for practice of pharmacy and REMS
Tocilizumab (Actemra®), anti-IL-6 receptor monoclonal antibody is available as a preservative-free 20 mg/mL single-dose solution individually packaged in 4 mL, 10 mL, and 20 mL vials, which need to be refrigerated at 2–8℃ (36–46°F). 35 All sizes are priced similar at average wholesale price (AWP) of $125.04. 37 The selection of vial sizes should be driven by the demographics of the served patient population and availability of shelf space. Minimizing the number of vials used for the preparation reduces the risk of a dispensing error and decreases the waste. The tocilizumab vials should be stored at a different location than prefilled syringes, which contain 162 mg of tocilizumab and are approved for subcutaneous administration in out-patient adults with rheumatoid arthritis and giant cell arteritis. Separating the storage locations can decrease potential for a dispensing error.
Since IL-6 reduces the CYP450 activities, counteracting IL-6 with tocilizumab can lead to activation of CYP450 system with effect persisting for several weeks. 35 This impact should be taken in consideration for patients on concurrent medications that undergo CYP450-dependent metabolism e.g. opioids, antidepressants, etc. Close monitoring with dose adjustments might be needed. Pharmacy staff need to be educated regarding location, preparation, and timely dispensing of tocilizumab especially during the off-hours, vacation, and holiday times. Reminders including relevant contacts might be added to Pharmacy Order Entry System to optimize drug dispensing. For those instances when tocilizumab is stored in refrigerated automated dispensing cabinets (ADCs), enclosing a protocol might be helpful for nurses involved in drug preparation and administration.
The goal of CAR T-cell REMS is to ensure that all members of an interdisciplinary team who prescribe, dispense and administer the therapies are trained on the management of CRS, and that the institution has immediate access to tocilizumab and is certified to dispense it.7,8 Recertification is required if institution did not dispense immunotherapy for a year or a new authorized representative is appointed. Conditions of attestation include designation of an authorized representative overseeing REMS compliance, staff including the authorized representative completing assessment of knowledge of CRS identification and treatment, and verification of availability of at least two doses of tocilizumab on site for each patient treated with immunotherapy for administration within 2 h.
As the best practices for grading and management of toxicities6,23,27,38 continue to evolve, the lack of standardized protocols might represent a challenge for practitioners with newly instituted programs. It appears that the approach proposed by Neelapu et al. 6 is suitable for adult patients, while the grading system and corresponding protocol suggested by Porter et al. 27 and Teachey et al. 23 is for the pediatric patient population. In addition, the use of both immunotherapies within one organization might further complicate the development of algorithms and order sets. Oncology pharmacist, medication safety officer, or other pharmacy practitioners involved in implementation of CAR T-cell programs need to collaborate with members of the team to resolve this patient safety matter. Adoption (with as needed modifications) of the order sets suggested by Neelapu et al. 6 might be helpful in tackling the issue. For organizations with computerized order entry (COE), incorporation of clinical decision support rules such as e.g. setting up the maximum allowed number of doses of tocilizumab in 24 h or maximum dosing limits for steroids and tocilizumab might be also beneficial in advancing patient safety and outcomes.
Conclusion
CAR T-cell therapy is a novel immunotherapy with promising outcomes for patients with hematologic malignancies. Three generations of CAR constructs had been developed; of those, two second-generation CAR T-cell immunotherapies (tisagenlecleucel, Kymriah™ and axicabtagene ciloleucel, Yescarta™) were approved. CAR T-cell therapies have been proved to be successful, at least for CD19-targeted lymphocytic malignancies. However, the adverse effects associated with the CAR T-cell therapy such as CRS might be severe and fatal. REMS for both Kymriah™ Yescarta™ is intended to mitigate the risks. Availability and appropriate administration of tocilizumab (Actemra®) for severe CRS is the core responsibility of pharmacists who are indispensable members of interprofessional patient care teams. Successful resolution of CRS and patient’s recovery require close coordination between all disciplines and extensive education of providers and patients.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.