
Abstract
Blinatumomab is the first in its class bispecific T-cell engager monoclonal antibody, which binds to CD19 expressed on B-cells and CD3 expressed on T-cells, resulting in lysis of CD19-positive cells common in B-cell malignancies. Blinatumomab is Food and Drug Administration (FDA) approved for the treatment of adults and children with relapsed/refractory or minimal residual disease (MRD) positive precursor B-cell ALL (B-ALL). Despite impressive efficacy for the approved indications and favorable toxicity profile compared to standard-of-care chemotherapy, blinatumomab presents unique health-system challenges related to preparation, administration, toxicity monitoring and medication error prevention. Blinatumomab delivery also offers plethora of opportunities for interdisciplinary planning and collaboration. The purpose of this paper is to discuss practical considerations for safe blinatumomab delivery from the pharmacy and nursing perspectives.
Background
Acute lymphoblastic leukemia (ALL) is the most common cancer in children younger than 15 years old. Globally, about 160,000 children and adolescents are diagnosed with ALL each year, with around 3,000 of these in the United States.1,2 In many countries, cooperative group clinical trials using multi-agent chemotherapeutic regimens have led to significant gains in survival over the past 40 years. Today, more than 85% of children diagnosed with ALL can expect long-term survival from their disease.1–3 Unfortunately, treatment of relapsed ALL is a more challenging scenario, and long-term cure following a relapse is less common. 4 In fact, 5-year overall survival from relapsed ALL is less than 40%. 5 Identifying therapeutic options to prevent and treat relapsed ALL are a high priority for the pediatric oncology community.
Targeted therapies that leverage and harness the immune system to treat malignancies have become one of the most rapidly growing areas in the field of oncology. Immunotherapy allows for a targeted approach to disease using novel mechanisms of action. Blinatumomab is one such agent, which has generated significant momentum in the treatment of ALL. It is a bispecific T-cell engager (BiTE), a single-chain protein with murine anti-CD19 and anti-CD3 antibodies. Blinatumomab exerts its anti-leukemic effects by binding to CD3 and engaging these T-cells to selectively target the CD19 antigen on B-cells. Early clinical trials of blinatumomab in relapsed and refractory ALL in adult patients were promising. In fact, the drug was granted accelerated approval by the FDA in December 2014, full approval in July 2017 for adults and children with relapsed/refractory B-ALL and an accelerated approval in March of 2018 to treat adult and children with B-ALL who have MRD greater than or equal to 0.1%. Subsequent clinical trials in adult patients have shown blinatumomab to nearly double rates of complete response, median overall survival, and 5-year event-free survival.6–8
The FDA approval of blinatumomab for pediatric relapsed/refractory Philadelphia chromosome (Ph) negative B-ALL was based on data from a Phase 1/2 international cooperative group trial of 70 patients less than 18 years of age (ClinicalTrials.gov Identifier: NCT01471782). 9 Of all relapsed and refractory patients who received the target dose of blinatumomab, CR was achieved by 39% within two cycles; for relapsed patients this was as high as 48%. More than half (52%) achieved MRD negativity, signaling a significant clinical benefit for the novel agent. 9 More recently, a Phase 3 clinical trial by the Children’s Oncology Group (COG) AALL1331 (ClinicalTrials.gov Identifier: NCT02101853) randomized patients with high and intermediate risk (HR/IR) B-ALL in first relapse to a standard re-induction backbone followed by either two blocks of chemotherapy or two cycles of blinatumomab for post-induction therapy. The randomization of this study was closed early when it was clear that the blinatumomab arm offered both significantly less toxicity and greater efficacy than the chemotherapy arm. 10 Although blinatumomab offers a reduced risk of toxicity compared to intensive chemotherapy in the post-induction phase for relapsed/refractory patients, it is not without significant toxicities of its own. The delivery of this agent is relatively complex and presents a variety of issues that must be carefully considered by the healthcare team. The purpose of this manuscript is to describe critical issues and recommendations for the delivery of blinatumomab to pediatric patients from the pharmacy and nursing perspective. Herein we highlight opportunities for interdisciplinary collaboration to deliver high quality of care, optimize the patient experience, and minimize the risk of errors or adverse events.
Delivering high quality care
Dosing
Blinatumomab is approved for pediatric patients with two dosing approaches: one for relapsed/refractory B-cell ALL and the other for MRD positive ALL. Each dosing approach includes a premedication approach, hospitalization when initiating therapy, and treatment-free intervals between cycles. Refer to Table 1 for additional details by indication. 11 Each patient should be carefully monitored and adjustments in the infusion rate or dose should be made based on toxicity-related dose adjustments (see: Adverse Event Management below).
Blinatumomab dosing in pediatric patients.
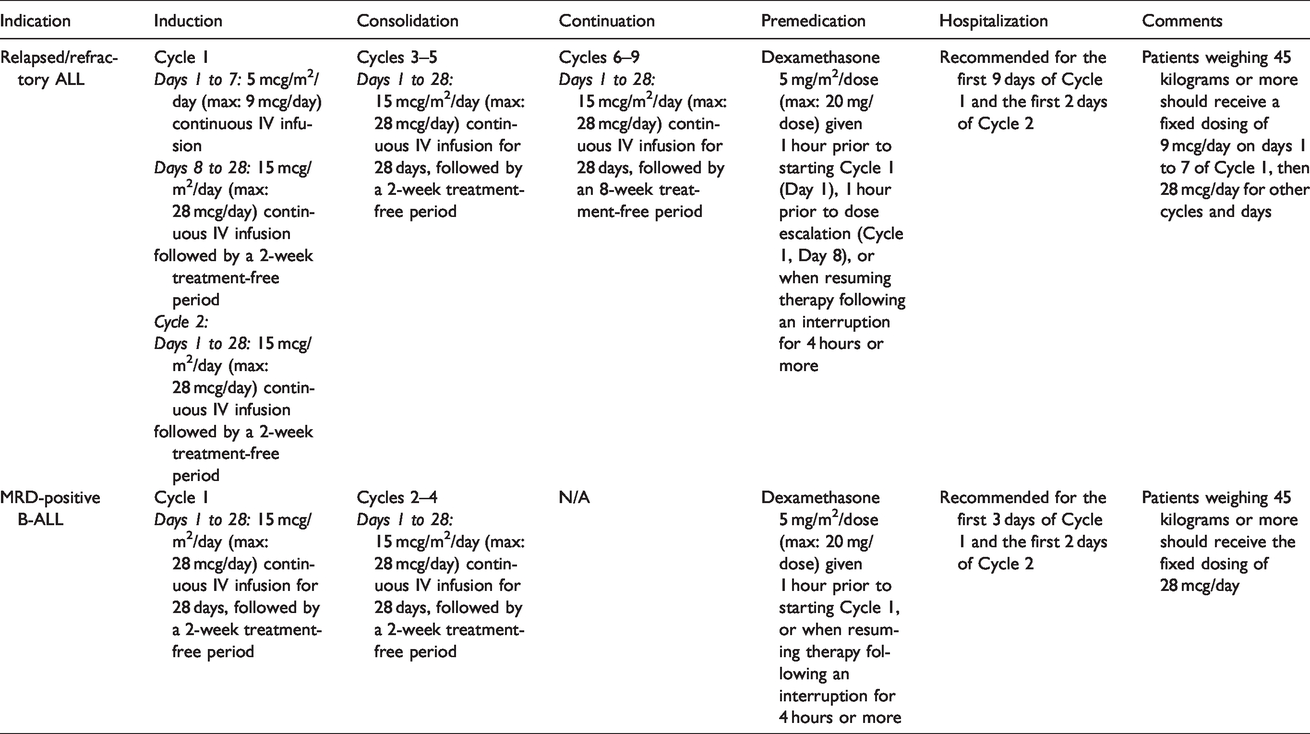
Although the dosing approach may seem straightforward, implementation of the dosing into contemporary electronic health records (EHRs) for computerized provider order entry is more complicated. Typically, EHRs are designed around more commonplace practices: oral and topical agents, injections, intermittent IV infusions, or for continuous infusions, standard “off the shelf” or custom IV fluids. The continuous infusions of IV fluids are typically built in units such as “mL/hour” or “mL/m2/hour.” They are not designed to be dosed in micrograms/m2/day, given continuously without stop, for 28 days, with 30 mL of overfill built in (but not delivered). Even with work-arounds for the dosing, determining the best way to build in overfill can be a challenge. To prevent overdose error and to decrease confusion, some institutions forbid the use of overfill. Others may elect additional work-arounds, such as systems which mandate the use of redundant paper forms for additional safeguards, manual re-entry processes at order verification or validation, conversion factors hard-coded to determine the dose with overfill, and medication label of electronic health record notes to indicate the delivered dose. None of the solutions offer a completely safe approach, and the intended dose, or even the actual delivered dose, could be easily misunderstood for persons not familiar with the system or labeling practices.
Preparation and dispensing
The prescribing information for blinatumomab includes specific instructions for preparation, dispensing and administration. Each bag is prepared for an intended duration of time. The U.S. prescribing information for blinatumomab includes 3 distinct approaches for the preparation of blinatumomab: 24-hour (10 mL/hour), 48-hour (5 mL/hour), and 168-hour (7-day; 0.6 mL/hour) infusions. 11 In other countries, 72-hour (3.3 mL/hour) and 96-hour (2.5 mL/hour) preparations are approved for use.12,13 Notably, the 7-day infusion bag approach cannot be used in children weighing less than 22 kilograms, as the final prepared product would deliver an excess (and unsafe) dose of benzyl alcohol over the duration of the infused dose. All materials used for dispensing (i.e., infusion bags, cassettes, IV tubing) must be made of polyolefin, ethylene vinyl acetate (EVA), or non- Di-2-ethylhexyl phthalate polyvinyl chloride (non-DEHP PVC). DEHP is a phthalate used to improve the flexibility of various plastics. DEHP has come under scrutiny in recent years due to its potential association with adverse health risks. Pharmaceuticals such as blinatumomab, particularly when given as a continuous infusion, may increase the risk of DEHP leaching from the plastic and entering the bloodstream. The details of these preparation methods and published stability data are described in Table 2.
FDA-approved blinatumomab dose preparation details.

aOnly non-DEHP PVC, EVA, or polyolefin bags or cassettes are permitted; products not meeting these criteria may result in leaching of DEHP into the solution.
Based on the number of manipulations, the materials used in the preparation of the final product, and the target patient population, blinatumomab may be considered a medium risk preparation when assigning beyond use dating according to USP <797>. 14 Under current official USP <797> guidance, a medium-risk agent such as blinatumomab (using sterile ingredients, but no sterility testing post preparation) may be assigned a beyond use date of 30 hours at controlled room temperature, or 9 days in a refrigerator. Proposed revisions to this guidance would allow for 96 hours at room temperature and 10 days in the refrigerator. 15 Using the preparation approaches included in the U.S. prescribing information (i.e., 24- and 48-hour bags), children must have access to home health/infusion services or an institution with services on the weekend to exchange the bag; neither of these options are routinely available in many settings. Moreover, the necessity for these services may add an undue hardship for parents or other caregivers or may widen disparities for patients without access or means to afford a higher, more complex level of care. Longer beyond use dating options approved in other countries allow for a longer duration of infusion (i.e., up to 96 hours),12,13 allowing patients to go home over the weekend, reducing operational strain on the institution, and decreasing the risk of infection related to the total times the patient’s line must be accessed during the course of the treatment cycle. For example, a patient weighing 20 kilograms will require a minimum of 14 bag changes per cycle when following the approved package labeling, compared to only 7 when using the methodology in pediatric clinical trials. Given the typical weight-for-age of children with newly diagnosed with ALL, the vast majority will not be eligible for 7-day bags and will require frequent bag changes (i.e., every 24 to 96 hours). Shortening the option for infusion duration from a maximum of 96 hours to a maximum of 48 hours is unnecessary, potentially harmful, and should be reconsidered.
Lastly, blinatumomab was added to the proposed draft of 2020 Hazardous Drug List from the National Institute for Occupational Safety and Health (NIOSH) due to its risk for neurotoxicity at extremely low doses. 16 The agent falls under Table 2, meaning that it is a drug that meets the NIOSH definition of a hazardous drug, but is not known to be a human carcinogen. Each institution will need to evaluate the risks of blinatumomab to determine their internal recommendations for the safe handling of the agent by healthcare providers, patients, and caregivers. 17
Administration
Infusion pumps
Planning for administration of blinatumomab to pediatric patients with B-ALL includes careful selection of the infusion pump. The selection of infusion pump must consider the infusion rate and dosing, the setting for the infusion (inpatient or outpatient administration), and the safety for the patient. For blinatumomab, the pump must be programmable, and have precise flow control using electronic infusion devices to ensure the accurate dose is delivered as prescribed. An elastomeric device is not appropriate. The infusion pump must have a lockable feature so that inadvertent dose changes cannot be made with manipulation of the pump. The choice of infusion pump for inpatient administration of blinatumomab will be influenced by individual site availability of infusion pumps. It may be necessary to use inpatient infusion pumps for any patient receiving therapy in the inpatient setting. If the treating institution will be renting outpatient infusion pumps or utilizing home health services after the patient is discharged, it may be necessary to administer blinatumomab using an inpatient infusion pump. Inpatient infusion pumps are not designed for low continuous infusion rates such as 5 mL/hour or less. In the situation when an inpatient infusion pump is used, blood backing up in the line and pump alarms indicating resistance are common. If this occurs, it might be necessary to run a companion solution that is compatible with blinatumomab (such as normal saline) to increase the total volume infused and eliminate blood backing up in the line.
The use of outpatient infusion pumps for administration of blinatumomab during an inpatient stay is an alternative option. Advantages of this approach include elimination of additional companion fluids, standardization of pumps for blinatumomab administration, and the opportunity for patients and families to become familiar with outpatient infusion pumps prior to hospital discharge. 18 A pump “sign-out” procedure or contract may help with tracking assignments of loaner pumps to specific patients. Outpatient infusion pumps can have a slight variance in delivery, around ±6%, which may cause the infusion to finish earlier or later than anticipated. Meticulous planning between nursing and pharmacy colleagues is essential to avoid gaps in the infusion of the drug. The inclusion of an overfill volume, as described in the blinatumomab prescribing information, may allow for a small buffer to help prevent these gaps. Additionally, infusion pumps must be programmed to allow for continued infusion of the reservoir volume, which exceeds the planned hours of infusion. It is crucial to inform families of this potential variation in time of blinatumomab delivery. Patients and families having scheduled blinatumomab done at an infusion center or treating hospital should be prepared to arrive ahead of scheduled blinatumumab bag changes to allow for this infusion variance. For unexplained and unacceptable variance in drug delivery (outside of the expected range for the pump), the infusion pump should be sent for assessment by biomedical engineering, and an alternate infusion pump should be used to administer the subsequent bag. For these reasons, many institutions have designated “back-up” pumps, or an extra pump per patient so an alternate pump is always available.
There is limited evidence to support strategies to minimize variation of outpatient pump infusions. However, it is suggested to maintain the infusion pump at the same height as the patient whenever possible to minimize the impact of gravity changes on the infusion rate, to maintain room temperature and avoid variation in temperature of infusion, and to assess the addition of any extra or add-on components (i.e., filters, needless access connectors) in the infusion set that might be interfering with the rate of infusion. One example of an extra component that may be contributing to variance in infusion rate is the addition of a needless access connector. Individual institutions will need to consider the risk and benefit of removing extra components, such as needless access connectors and closed system drug transfer devices (CSTD), considering the importance of supporting accurate dose delivery of blinatumomab. If these extra components are removed to minimize variance, safety of the frontline staff involved in administration will need to be considered and there may be a need for additional personal protective equipment when preparing and administering blinatumomab.
Infusion sets/tubing
As with the preparation and dispensing process, blinatumomab should not be administered through tubing sets or components containing DEHP. Only non-DEHP PVC, polyolefin, or EVA tubing/lines with a 0.2 µm inline filter are acceptable for bags prepared for 24 to 96-hour infusions. For 168-hour (7-day) infusions prepared with bacteriostatic sodium chloride, an in-line filter is not required. These tubing sets may require special ordering procedures through a designated supplier if not readily available through the hospital’s regular supply chain process.
Securing devices and extension sets
Consideration of best methods to secure the infusion sets and tubing in younger pediatric patients will be important to prevent dislodgment and/or disconnections. The central line dressing for the Central Venous Access Device (CVAD) alone is unlikely to provide sufficient securement of the infusion set and tubing in an active patient for the entire 28-day cycle of blinatumomab. Securement of the tubing set with an engineered securing device (ESD) is recommended. 18 Other methods of securing the tubing set will require careful decision-making around patient risk and benefit. Adhesives and tapes applied directly to the skin have not been found effective in improving securement and can introduce a risk of skin breakdown and subsequent infection. 19
It is important to allow for movement and mobility of the pediatric patient receiving a 28-day continuous infusion cycle and this could be accomplished with the addition of an extension tubing set. Extension tubing sets can be advantageous in minimizing movement and manipulation at the site of the catheter hub, however tubing extension sets add additional risk for disconnection and misconnection, and methods to secure extensions will be important. If using an extension tubing set, ensure it is compatible with blinatumomab administration (i.e., DEHP-free).
Line flushing
Most pediatric patients with B-ALL will have a CVAD placed at the time of initial leukemia diagnosis or at the time of relapse, and placement before initiation of blinatumomab is recommended. The CVAD will be either an external line such as a peripherally inserted central catheter (PICC), cuffed tunneled vascular access device (CVAD) or an implanted vascular access port (PORT). Flushing venous access devices, including central lines, is crucial to ensuring the line is patent and functional, and to prevent complications that can be harmful to patients. 20 Typically, it is recommended to flush CVADs after each infusion and lock after completion of the infusion to minimize the risk of intraluminal occlusion and catheter-related bloodstream infection.21–23 However, intermittent flushing of lines is a potentially dangerous practice during a cycle of blinatumomab due to the risk of inadvertent bolus of blinatumomab. For example, a single flush with a typical flush volume (5 to 10 mL) could deliver a one hour dose of blinatumomab over several minutes. As such, attempts to limit flushing the line are recommended where possible. For example, it is not necessary to “check” the line daily to confirm CVAD placement with blood return, a procedure which requires the line to be flushed. The practice for flushing the line during blinatumomab administration should be reviewed by each institution.
Interruptions of the continuous infusion
It is important to minimize interruptions to the blinatumomab infusion over the continuous 28-day cycle to optimize the dose delivered. However, some interruptions will be necessary. For pediatric patients with a PORT, needle changes will be required on a schedule detailed by each institution’s internal policy. Most pediatric oncology patients with PORTs will require a needle change at minimum every 7 days. Some institutions have made a standard operating procedure (SOP) to deviate and allow for PORT needle change every 8 days to align with blinatumomab bag changes. The process for PORT needle change can result in an hour or longer interruption to allow for application of local anesthetic creams afterwards to minimize needle stick pain with re-access. An hour to allow for full benefit of anesthetic creams is recommended. Careful planning around required PORT needle changes can help to minimize the time of interruption of the blinatumomab infusion. PORT needle changes will require a new bag of blinatumomab with a new infusion/tubing set. Nursing and pharmacy should work closely to ensure that a newly prepared blinatumomab bag and primed tubing set are available and ready to hang immediately prior to reinsertion of the PORT needle to maximize the amount of time the bag can be used and minimize the gap of time between removing one bag and initiating the next. A second infusion pump can be used to prime the new infusion set, allowing this process to be done while the current bag continues to infuse, thereby minimizing interruptions.
Infusion interruptions will be necessary for other clinical care needs, such as blood draws, administering other medications and supportive care therapies, and to provide sedation for painful procedures. Institutions will need to develop a process to track interruptions to the blinatumomab infusion that are greater than one hour and to determine cumulative interruptions during a 28-day cycle. This will be necessary to capture cumulative interruptions that would be considered significant and thus impact the total dose administered per cycle of therapy. Total time of interruption that would warrant a significant dose discrepancy is not established. If a patient is registered on a clinical trial, the specifications of the trial should be followed for any “make-up” dose at the end of the cycle. For patients receiving commercial blinatumomab not on a clinical trial, the decision for the total time of interruption and need to make-up the dose should be made on an individual case basis with the treating oncologist.
Scheduling day 1 of each cycle
Timing the start of a blinatumomab cycle will be an important consideration. The start time will dictate the timing of subsequent bag changes, based on 24, 48, 72, 96, and 168-hour bag preparations. The U.S. prescribing information for commercial blinatumomab only provides stability and preparation information for 24 hour, 48 hour and 168 hour bag changes. 11 Patients registered on clinical trials receiving investigational blinatumomab supply may have additional options of 72-hour and 96-hour bag changes (e.g., COG AALL1731 trial; ClinicalTrials.gov Identifier: NCT03914625). The infusion start time on day 1 should be scheduled to take into consideration subsequent bag change times. For example, if the patient will go home during the second week of therapy, the infusion should be scheduled during clinic business hours to prevent drug waste or interruption of the infusion due to insufficient product dispensed for the intended dosing time frame. Advanced preparation for admissions is essential; prescribing, reviewing and timing medication orders in advance can prevent delays. Team huddles to plan for nursing assignment and patient needs (e.g., clinical supplies) can also prevent delays in start of the infusion on day 1.
Nursing assignments/nurse to patients ratio
There is no evidence to support the optimal nurse to patient ratio for patients receiving inpatient blinatumomab, particularly for the first cycle. Although rates of grade 3 and 4 cytokine release syndrome (CRS) and neurotoxicity have been low in pediatric clinical trials with blinatumomab, there is still a risk of these serious side effects that will require intensive nursing monitoring and support. 10 It is therefore important to have appropriate nursing staff assigned to the patient receiving blinatumomab to allow for potential escalated care needs. The greatest risk of both CRS and neurotoxicity has been seen in patients with first exposure, or first cycle of blinatumomab. 24 There is no current evidence to support the frequency of vital signs and assessment or neuro-vital signs for patients receiving the first cycle of blinatumomab. A conservative approach to vital signs, with an intensive frequency for monitoring, and a limited nurse to patient ratio should be employed. Many centers have adopted the practice of alerting their intensive care colleagues when a patient is planned to receive their first cycle of blinatumomab given the potential need for ICU level care.
Optimize the patient experience
Education of patients and families
Most of a blinatumomab cycle can be administered in the outpatient setting. There are general principles for providing education to parents and caregivers that include: proper care of the child’s CVAD, precautions for preventing infection when providing care related to the infusion or CVAD, signs and symptoms that require reporting or urgent attention from a member of the medical care team, and where and how to report concerns including these signs and symptoms. Parents and caregivers will need education on the care of the electronic infusion device, specifically the administration pump, how to troubleshoot alarms of the infusion pump, and who to contact for infusion and pump related concerns. Careful consideration must be taken for infusion pump troubleshooting support during off hours (evenings, weekends, and holidays) to ensure that caregivers have quick and easy access to practitioners familiar with the infusion device. While the emergency department may be the usual recommendation for care required during off hours, familiarity and competency with the infusion pump may be limited. Some institutions have developed a procedure/process that parents/caregivers can directly call the inpatient oncology unit during off hours and speak with a nurse who is familiar with the outpatient infusion pump. The nurse will need to discern whether the concern is directly related to the infusion pump, or if there are clinical concerns for the patient receiving the blinatumomab and appropriately triage. The main advantages to this approach are quick access for families with potential over-the-phone solutions that will minimize interruptions to the infusion and avoid a return trip to the treating center or emergency department.
Another strategy to support patient safety during outpatient administration of blinatumomab is to provide education to parents and caregivers specific to the anticipated alarms with the intended infusion pump. An end-user tip sheet can be developed to help trouble shoot these specific alarms. The tip sheet can be provided to the parent/caregiver together with verbal (in-person) review by a nurse familiar with the infusion pump and its potential alarms. At the time of each bag change, the clinical team should take advantage of the opportunity to review the user tip sheet, any alarms that may have or could potentially occur, as well as who, how and when to contact the care team. Institutions should consider how they can document the education provided prior to the first discharge and at each clinical interaction, in-person or by phone, with the patient/family/caregivers. A policy for documentation will ensure appropriate education has been provided. It will be important to evaluate the parent/caregiver understanding of education provided. Methods to evaluate parent/caregiver understanding can include verbal feedback of information (teach-back) and demonstration of psychomotor skills (handling of pump and alarms). This should occur prior to the first discharge to ensure family readiness and patient safety once in the outpatient setting.
Distance to hospital/treating center
During early phase trials with blinatumomab in the treatment of pediatric relapsed B-ALL the clinical course and toxicities were unknown. For patients enrolled on early phase trials it was strongly recommended that patients/families stay in close distance, recommended no more than one hour anticipated travel time to the hospital, to allow for quick access to medical assessment and care. This was necessary to ensure both patient safety with a new agent and to capture all necessary data for the early phase trials. There is no current evidence-based guidance on a recommended safe distance or travel time to the hospital for patient’s receiving blinatumomab in the outpatient setting. A risk based approach to this decision will need to be made for each individual patient and family considering important factors such as ability to return to the hospital, including appropriate transportation, perceived parent/caregiver competence to provide care in the home setting, perceived parent/caregiver knowledge and understanding of when and how to call for help. Specific to blinatumomab would be the consideration of length of anticipated interruption if the infusion would require stopping until the patient reached the hospital for assessment.
Adverse event management
The side effect profile of blinatumomab reflects its mechanism of action involving T-cell activation and subsequent cytokine release, with most significant toxicities being CRS and neurotoxicity. In general, blinatumomab-related adverse events are similar between pediatric and adult patients, with certain adverse events (fever, hypertension, anemia, infusion-related reactions, thrombocytopenia, leukopenia, and weight increase) occurring at a higher frequency in pediatric patients. 11 Infants also had a higher incidence of hypokalemia compared to older children and adults. 11
Cytokine release syndrome (CRS)
CRS is a systemic inflammatory response resulting from antigen-antibody interaction leading to the release of inflammatory cytokines into the blood stream. IL-10, IL-6 and INFγ appear to be the most highly elevated cytokines after blinatumomab treatment. 25 The reported incidence of all-grade CRS in pediatric and young adult patients with relapsed/refractory ALL is 22% based on the recent results of Children’s Oncology Group AALL1331 study that enrolled 208 patients between 1–30 years of age. 10 The exact mechanism remains unclear, however, it has been proposed that patients with more severe forms of CRS may experience abnormal macrophage activation triggered by inflammatory cytokines release leading to hemophagocytic lymphohistiocytosis (HLH)-like picture. 24 IL-6 is thought to be a good target to mitigate toxicity of CRS without compromising T cell-mediated anti-leukemic activity.
CRS consists of a constellation of clinical symptoms ranging from mild (fever, malaise, headache, nausea, skin rash or myalgias) to life-threatening and may be indistinguishable from other infusion-related reactions or an infectious process. Severe CRS can result in capillary leak, hypotension, hypoxia, pulmonary edema, coagulopathy, transaminitis, hyperbilirubinemia, renal impairment, cardiac dysfunction, multiorgan system failure and death. 11 Most CRS events usually occurs within the first 7 days of the first blinatumomab cycle (median time to onset of 2 days) with a median time to resolution of 5 days in cases that resolved. 11 It has been suggested that higher disease burden and higher initial dose of blinatumomab are associated with higher risk of CRS, with reported incidence of up to 22% in patients with relapsed/refractory ALL.10,11
In order to mitigate the risk of CRS, several prevention strategies have been implemented. In patients with relapsed/refractory B-ALL where the disease burden is expected to be high, the step up dosing of blinatumomab is utilized during the first cycle of therapy. For adults, the recommended blinatumomab step up dosing is 9 micrograms/day IV continuous infusion Days 1–7 of therapy, followed by 28 micrograms/day on Days 8–28 and on subsequent cycles. For pediatric patients, the equivalent step up dosing during Cycle 1 is 5 micrograms/m2/day (maximum 9 micrograms/day) during the 1st week, followed by 15 micrograms/m2/day (maximum 28 micrograms/day) Days 8–28 and on subsequent cycles. In addition, premedication with dexamethasone (5 mg/m2/dose up to a maximum of 20 mg) is required one hour prior to the first dose of blinatumomab, prior to the step up dose (Cycle 1, Day 8) and when restarting infusion after an interruption of 4 hours or more to minimize occurrence and severity of CRS and infusion reactions. 11 Patients should be closely observed for CRS and other toxicities, hence hospitalization is recommended for the first 9 days of the first cycle and the first 2 days of the second cycle in patients with relapsed or refractory ALL. B-ALL patients with minimal residual disease appear to be at a lower risk of CRS and should be hospitalized for the first three days of the first cycle and the first two days of the second cycle. For all subsequent cycle starts and re-initiation (e.g., if treatment is interrupted for 4 or more hours), supervision by a healthcare professional or hospitalization is recommended. 11
Several CRS grading systems have emerged since development of blinatumomab and chimeric antigen receptor therapy T-cell (CAR-T) cell therapy in an effort to standardize approach to CRS management.26–29 Table 3 outlines the differences between the current National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0 and American Society for Transplantation and Cellular Therapy (ASTCT) CRS grading systems.27,29
Comparison of CTCAE and ASTCT CRS grading systems.

aFever is defined as temperature ≥38°C.
bLow-flow nasal cannula is defined as oxygen delivered at 6 L/minute. Low flow also includes blow-by oxygen delivery, sometimes used in pediatrics. High flow nasal cannula is defined as oxygen delivered at >6 L/minute.
The ideal CRS grading system should have broad applicability regardless of product used (e.g., CAR-T vs monoclonal antibody therapy) and have well-defined criteria to guide therapeutic decisions. ASTCT grading system was developed in an attempt to harmonize disparities among previously published grading systems. The choice of CRS grading criteria is often left up to the treating clinician or specified in the investigational protocol. As you can see from Table 3, ASTCT criteria have more specific CRS grading definitions compared to CTCAE criteria that may be helpful in guiding therapy selection, especially with the trend of earlier use of anti-cytokine therapy allowing for earlier resolution of CRS while still preserving efficacy of blinatumomab.
Management of CRS depends on CRS grading and may require temporary infusion interruption or discontinuation of blinatumomab treatment. Due to a short half-life of blinatumomab (2.1 hours) 11 these events may resolve relatively quickly after infusion interruption. Close monitoring of fluid status is critical for the first 48 hours of blinatumomab infusion. Efforts to keep patients balanced between intake and output should be maintained, even if diuretic therapy is needed to do this to prevent capillary leak syndrome and subsequent deterioration. Tocilizumab is a recombinant humanized monoclonal antibody directed against both soluble and membrane bound IL-6 receptor, which may result in rapid clinical stabilization of CRS. Tocilizumab is FDA approved for the treatment of severe or life-threatening CRS in children 2 years of age or older and adults following CAR-T therapy. 30 The recommended dose of tocilizumab is 12 mg/kg for patients <30 kg of weight and 8 mg/kg (maximum 800 mg) for patients ≥30 kg of weight administered intravenously over 1 hour. Fever and tachycardia typically resolve within hours of tocilizumab administration; however, resolution of hypotension and organ dysfunction is delayed. If no clinical improvement occurs after the first dose, up to 3 additional doses of tocilizumab at least 8 hours apart can be administered. Timing of tocilizumab administration is a clinical decision based on CRS severity grading and circulating cytokine levels. Serum IL-6 measurements are not readily available outside of the clinical trial setting, hence C-reactive protein (CRP) levels are often used as surrogate markers of CRS onset and progression, albeit non-specific and often elevated by infectious or inflammatory causes. Presence of fever is a pre-requisite for initiation of anti-cytokine therapy. Other medical conditions such as infection, febrile neutropenia, tumor lysis syndrome, or adrenal insufficiency should be ruled out prior to tocilizumab administration. A second line agent such as corticosteroid should be introduced if CRS fails to resolve after the first dose of tocilizumab. Siltuximab, is another anti-IL-6 chimeric monoclonal antibody that has been utilized by some centers if tocilizumab and steroids fail to improve CRS. 31 The use of siltuximab in CRS has not been sufficiently studied and is deemed investigational. CRS treatment algorithms may vary institutionally, however, the key components include interruption or discontinuation of blinatumomab infusion, use of supportive care agents such as antipyretics and analgesics, empiric treatment for febrile neutropenia, close monitoring of fluid status and organ functions, initiation of tocilizumab +/− corticosteroids for patients with CRS grade 2 or higher depending on co-morbidities.
Neurotoxicity
Neurotoxicity is another common toxicity observed with blinatumomab. Neurotoxicity was initially considered an aggregate of CRS, but was later recognized to be a separate entity. The mechanism of blinatumomab-induced neurotoxicity is not fully elucidated, but may be related to disruption of blood brain barrier by activated T-cells and the release of neurotoxic cytokines in the central nervous system (CNS) causing inflammation of neuroendothelium. Headache is commonly seen in patients receiving blinatumomab (39%) 11 but it is not representative of neurotoxicity specifically. Neurotoxicity often manifests in tremor, cognitive disturbance, ataxia, dysarthria, seizures, aphasia, difficulty concentrating, lethargy, delirium, agitation, encephalopathy and rarely cerebral edema. Manifestation of neurologic toxicity and its monitoring may depend of the age of the child. In pediatric patients younger than 2 years old, the incidence of neurologic toxicities was not significantly different than in older age groups, but its manifestations may differ with irritability, somnolence, agitation, insomnia and headache reported most commonly. 11
Neurologic symptoms can occur at the same time or after the onset of CRS. They rarely occur before CRS onset. Neurotoxicity is usually rapidly reversible upon discontinuation of blinatumomab and often does not recur upon re-challenge. In COG AALL1121 trial (ClinicalTrials.gov Identifier: NCT01471782), 13% patients experienced neurologic events (mainly tremor and dizziness) that were considered treatment related. 9 All neurotoxicity events were Grade 2 and resolved, with no permanent discontinuations due to neurologic events. As a large monoclonal antibody, tocilizumab is not expected to penetrate the blood–brain barrier and its CSF concentrations are unknown. It would be unlikely to control CSF inflammation induced by blinatumomab or CAR-T cell therapy. Dexamethasone has been used to treat neurologic adverse events in some adult studies and can be considered. 32
Since most patients with ALL require CNS prophylaxis, the question of timing of intrathecal (IT) chemotherapy and blinatumomab often comes up. Some case reports suggested increased neurotoxicity with concurrent IT and blinatumomab administration. 33 IT therapy during blinatumomab blocks was administered in both COG AALL1331 trial and the currently enrolling COG AALL1731 frontline trial. There was no increase in neurological adverse events noticed in AALL1331 trial during blinatumomab blocks. 10
Other toxicities
Other common blinatumomab toxicities include cytopenias, abnormal liver function tests, and hypogammaglobulinemia. It’s not clear if decrease in IgG levels translates into increase in infectious complications with blinatumomab. COG AALL1331 study results actually demonstrated reduction in grade 3 or higher febrile neutropenia, infections, and sepsis in blinatumomab arms compared to conventional chemotherapy. 10 Periodic monitoring of immunoglobulin levels, as well as intravenous immunoglobulin (IVIG) replacement therapy in patients with severe infections and low serum IgG concentrations, may be considered. The most common grade 3/4 hematologic events are lymphopenia, leukopenia, thrombocytopenia, febrile neutropenia, and anemia.
Medication errors and risk evaluation and mitigation strategy (REMS)
Blinatumomab may be uniquely susceptible to medication errors related to drug preparation and administration due to complexities of product preparation requiring the use of solution stabilizer (to coat prefilled IV bag), the need for overfill, differences between commercial and investigational drug supply, variable final product volumes depending on infusion duration, variable infusion rates, requirement for microgram/m2/day dosing in children vs flat dosing in adults or patients weighting more than 45 kg, continuous infusion nature of drug administration and inherent pump rate variability. Strict adherence to preparation and administration guidelines combined with staff and caregiver education is required to prevent these errors from occurring. Sites treating pediatric patients should pay special attention to whether commercial supply vs investigational supply of blinatumomab is being used. Commercial blinatumomab is available in 35 microgram vials, whereas investigational supply provided for the purposes of clinical trials (e.g., COG AALL1731 protocol) contains 38.5 micrograms of blinatumomab per vial.
In addition, clinical trials utilizing investigational supply of blinatumomab often allow for additional infusion durations/bag sizes (e.g., 72 hour and 96 hour infusions), while U.S. prescribing information only provides stability and recommendations for 24-hour, 48-hour and 168-hours bags. These differences in infusion bags preparation and timing of bag changes can result in caregiver and provider confusion due to differences in rates of infusion (i.e., 10 mL/hour for 24-hour bags vs. 5 mL/hr for 48-hour bags) and may potentially result in under-dose or over-dose errors. To improve the safety of administration, reduce the risk of over- or underdosing, and optimize patient satisfaction, we urge regulatory agencies and the manufacturer to consider alternate preparation approaches which allow for a consistent rate of administration across all bag sizes. See the Figure 1 for medication safety and practical concerns in two example patient cases.

Patient cases for consideration.
A Risk Evaluation and Mitigation Strategy (REMS) is a drug safety program required by the U.S. Food and Drug Administration (FDA) for certain high-risk medications associated with potential serious safety issues in order to maintain the balance of the benefits outweighing the risks. Blinatumomab REMS contains three essential components: information on incidence, manifestations, and monitoring for CRS, neurological toxicity and avoidance of preparation/administration errors. 34 It also contains fact sheet and letter for healthcare providers as well as a letter for hospital and homecare pharmacists.
Conclusions
Blinatumomab is a novel targeted agent with an impressive efficacy profile for relapsed/refractory and MRD (+) precursor B-cell ALL in both pediatric and adult patients. Its complex preparation and administration process along with some targeted toxicities present a unique opportunity for collaboration among healthcare providers and requires careful planning and coordination. Communication between information technology personnel, oncologists, oncology clinical pharmacists and nurses is necessary for creating and testing of blinatumomab order sets in the EHRs for inpatient administration. Early identification of homecare agencies available to safely prepare and administer blinatumomab in the outpatient setting can allow for smoother transition from inpatient to home environment. For areas where homecare access is unavailable or limited by the lack of competency, resources and reimbursement, planning for blinatumomab bag changes in the outpatient oncology setting will need to take place to allow for sufficient staffing and provider/caregiver education. Pharmacy departments need time to implement a drug preparation process for both commercial and investigational blinatumomab use due to differences in the formulation (vial size) and preparation process. Nursing departments may need to discuss staffing levels, monitoring recommendations, plans to minimize infusion interruptions and line care, order necessary pumps and extension/tubing sets, and create both provider and caregiver education materials. Provider and patient/caregiver education is paramount to the success of safe blinatumomab administration as it relates to early recognition, prevention and management of CRS, neurotoxicity and medication errors. Despite the potential challenges, a number of pediatric cancer centers in US, Canada, and other countries have been successful in setting up a safe blinatumomab delivery program facilitating access to an immune therapy with potential evolving clinical applications.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.