
Abstract
Purpose
Lenvatinib is an oral multi-kinase inhibitor prescribed in renal cell carcinoma (RCC), endometrial cancer (EC), hepatocellular carcinoma (HCC), and radioactive iodine-refractory differentiated thyroid cancer (DTC). Lenvatinib is poorly tolerated at the recommended label dose (RLD), with > 60% of patients requiring dose modifications. Given that studies have identified an exposure–response relationship, therapeutic drug monitoring (TDM) has been proposed as a potential strategy to improve tolerability. This single-centre study reviews lenvatinib dosing across cancer types and discuss the potential role of TDM.
Methods
Medical records from a hospital in South Australia were retrospectively reviewed for patients treated with lenvatinib between December 1, 2016, and December 31, 2023. Data on cancer diagnoses, dose, and demographics were collected and analysed.
Results
Eighteen patients (19 treatment episodes), median age 61 years (range: 34–79), received lenvatinib. Cancer types included DTC (n = 10), EC (n = 4), HCC (n = 3), RCC (n = 1), and adenoid cystic carcinoma (n = 1). Lenvatinib was started at a reduced dose in 42% of the treatment episodes, and of these episodes 87% were never escalated to the RLD. At least one adverse event related to lenvatinib was reported in 95% of all treatment episodes during the study period; adverse events were identified from clinical documentation and were not graded using standardised criteria.
Conclusion
This study suggests variability in clinical practice relative to labelled recommendations for lenvatinib dosing. Even with initial dose reductions, adverse events and treatment interruptions still occur. Future studies should define target lenvatinib concentrations to facilitate clinical TDM application.
Introduction
Lenvatinib is an oral multi-kinase inhibitor used in the treatment of several solid tumours, including renal cell carcinoma (RCC), endometrial carcinoma (EC), differentiated thyroid cancer (DTC), and hepatocellular carcinoma (HCC). 1 It selectively targets a broad spectrum of receptors, including vascular endothelial growth factor receptors (VEGFR) 1–3, fibroblast growth factor receptors (FGFR) 1–4, platelet-derived growth factor receptor α (PDGFRα), stem cell growth factor receptor (c-KIT), and proto-oncogene tyrosine-protein kinase receptor (RET). 1
The recommended label dose (RLD) of lenvatinib is indication specific.2–5 For RCC and EC, the recommended dose is 20 mg/day, combined with pembrolizumab administered every 21 days for 35 cycles.2,3,6 Whilst it is given as a monotherapy in DTC (24 mg/day) 7 and HCC (8 mg/day for patients <60 kg and 12 mg/day for those ≥60 kg). 8 Although guidelines recommend continuing treatment as tolerated until disease progression, achieving adequate relative dose intensity (RDI) can be challenging with lenvatinib in clinical practice. Studies have reported that maintaining a high RDI of ≥60–70% early in the treatment cycle with lenvatinib is associated with favourable outcomes.9,10 However, achieving this target dose intensity is difficult in clinical practice due to the high incidence of treatment-related adverse effects.11,12
Lenvatinib is known to be poorly tolerated with studies reporting rates of dose reduction (> 60%), interruption (>60%), and/or discontinuation due to treatment-related adverse effects, across cancer types, when administered at the RLD.13–17 The median onset of lenvatinib-associated adverse effects is reported to be approximately 37 days. 18 Common adverse effects include, hypertension, fatigue, stomatitis, diarrhoea, and thyroid dysfunction. 18 This high incidence of toxicity creates a clinical challenge in balancing sufficient drug exposure with the patient's tolerance of therapy.
These real-world tolerability challenges contribute to significant variability in how lenvatinib is dosed in practice, prior studies found that clinicians relied on empirical dose reductions or planned treatment breaks to mitigate adverse events.19–22 Emerging evidence suggests that alternate approaches, such as therapeutic drug monitoring (TDM), may help guide dose individualisation for lenvatinib.13,23–25 Oncology pharmacists, who are central to patient education, dose review, and toxicity management, are key in advocating for and implementing precision dosing strategies like TDM to optimise dosing of cancer drugs.26,27 However, the role of routine TDM for lenvatinib and other cancer drugs is not yet clearly defined in clinical guidelines.
To better inform these dose optimisation strategies, a clear understanding of current practice is essential. The purpose of this single-centre retrospective study is to evaluate real-world dosing patterns, tolerability, and dose modifications across multiple cancer types. Given that lenvatinib is used across multiple tumour types with similar tolerability challenges, this study aimed to evaluate dosing practices across indications to better reflect real-world clinical use. We subsequently discuss the potential for oncology pharmacists to facilitate therapeutic drug monitoring (TDM) as a strategy for individualised lenvatinib dosing.
Methods
Study design and site
This retrospective study was conducted at the Flinders Medical Centre, a public tertiary hospital in South Australia. Ethics approval was granted by Southern Adelaide Clinical Human Research Ethics Committee, approval number 105.24. The study was classified as low risk, and the requirement for patient consent was waived in accordance with section 2.3.10 of the National Statement on Ethical Conduct in Human Research (2023). 28
Data sources and patient identification
Patients who received lenvatinib for any cancer indication between 1 December 2016 and 31 December 2023 were identified through oncology medical records and SA Health databases. Eligible patients were included if sufficient clinical, dosing, and treatment documentation was available. These patients were subsequently categorised by cancer diagnosis for further analysis.
Data collection and processing
Demographic variables, cancer diagnoses, treatment duration, dosing information, adverse events, comorbidities, concomitant medications, lenvatinib dosing details and relevant biochemistry were extracted for each patient. Data were collected by one author (JT) and independently reviewed by a second author (RH) to ensure accuracy.
Lenvatinib dosing in this study was reported as daily averages to facilitate comparisons, as some patients received intermittent dosing (e.g., seven days on and seven days off) instead of continuous daily dosing. All patients were assumed to be 100% adherent to their oral lenvatinib, unless if the clinical documentation specifies non-compliance.
Adverse events were identified based on clinician-documented notes in the medical record. Standardised toxicity grading criteria such as the Common Terminology Criteria for Adverse Events (CTCAE) were not retrospectively applied due to disparate documentation.
Baseline laboratory values were defined as those obtained within 28 days prior to lenvatinib initiation, or within 7 days after initiation when earlier values were unavailable. Baseline comorbidities and concomitant medications were determined from medical records; where documentation was incomplete, medication histories were used to infer comorbidities.
Treatment end dates were obtained from clinical documentation or when unavailable, estimated based on pharmacy dispensing records. Reasons for treatment discontinuation were determined from clinician-documented notes in the medical record where available, including attribution to adverse events, disease progression, or other clinical factors.
Relative dose intensity
Relative dose intensity (RDI) was calculated as the ratio of the cumulative lenvatinib dose received to the cumulative RLD for lenvatinib over the same period. Both overall RDI and 8-week RDI were estimated. A detailed explanation and formulae utilised to ascertain the RDI is available in Supplementary Table 1.
Outcomes and statistical analysis
The primary outcome of this study was to evaluate the dosing patterns of lenvatinib in patients treated for cancer. Descriptive statistics were used to summarise dosing patterns, toxicity events and dosing intensity. Given the small sample size, clinical heterogeneity of the cohort and non-standardised clinical documentation, inferential statistical analyses and formal group comparisons were not performed. A swimmer's plot was implemented to visualise the events of dose adjustments, toxicities and treatment breaks throughout the course of lenvatinib treatment for each patient. For patients continuing lenvatinib beyond the study period, treatment end dates were censored at the study end date.
All statistical analyses were performed using Excel and R version 4.4.1. 29
Results
Patient characteristics
During the study period, 24 patients received lenvatinib at the study site. Six patients were excluded from further analysis: five due to unavailable information and one because they were treated with lenvatinib for only two days before discontinuing treatment for palliative reasons. Cancer types included DTC (n = 10), EC (n = 4), HCC (n = 3), RCC (n = 1) and adenoid cystic carcinoma (n = 1). One patient with DTC had a treatment break exceeding one year due to adverse events and was treated as two separate episodes (Patient IDs: 5 and 5*), making a total cohort of 19 treatment episodes. Patient characteristics are detailed in Table 1 and Supplementary Table 2.
Demographics and baseline characteristics of patient treatment episodes treated with lenvatinib.

*Baseline laboratory data was not available for one of the patients with thyroid cancer Count includes a single patient (patient ID 5 and 5*) counted as 2 separate episodes for the purpose of analysis.
†Count includes a single patient (patient ID 5 and 5*) counted as 2 separate episodes for the purpose of analysis.
Dosing patterns
Across all cancer types, 11 of 19 (58%) treatment episodes were initiated at the RLD, while 8 episodes (42%) were commenced at a reduced dose (Table 2; Supplementary Table 3). Among patients that commenced at the reduced dose, 7 of 8 episodes (87%) were never escalated to the RLD at any time during the study period. Four patients received intermittent dosing schedules with the use of planned treatment breaks (Supplementary Table 3) during the follow up period.
Description of lenvatinib dosing patterns in the cohort of 19 patient episodes.
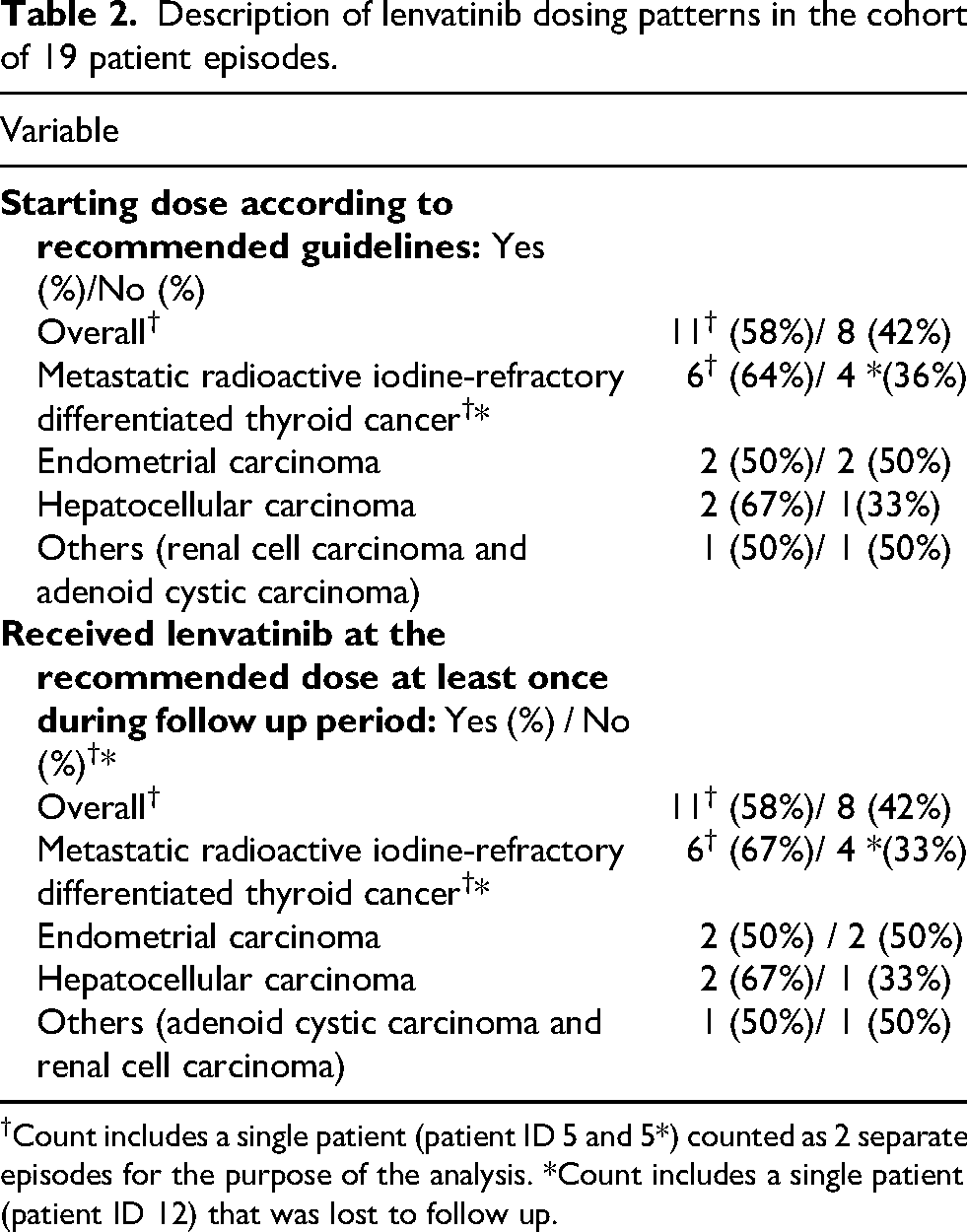
†Count includes a single patient (patient ID 5 and 5*) counted as 2 separate episodes for the purpose of the analysis. *Count includes a single patient (patient ID 12) that was lost to follow up.
Dose intensity
No patients received a lenvatinib dose exceeding the RLD for their cancer types. The median 8-week RDI for the overall cohort was 70% (IQR: 55–96). Patients who commenced at a reduced dose had a median 8-weeks RDI of 57% (IQR: 50–60), compared with 91% (IQR: 79–100) for those treated at the RLD (Table 3, Supplementary Table 3). Among patients that discontinued lenvatinib treatment within the follow up period, the median overall RDI was 62% (IQR: 46–80) (Table 3).
Description of lenvatinib dose intensity for patient episodes.

†Includes a single patient (patient ID 5 and 5*) counted as 2 separate episodes for the purpose of the analysis.
*One patient (patient ID: 12) was excluded due to loss of follow up, two patients (patient ID: 9 and 10) was excluded as they continued treatment beyond the follow up period.
Treatment tolerability and outcomes
18 of 19 treatment episodes (95%) experienced at least one adverse event attributed to lenvatinib during the study period (Figure 1, Supplementary Table 3). Adverse events were purely identified using clinical documentation and were not graded using standardised criteria such as the CTCAE. The swimmer's plot shows variation in treatment duration across patients and between cancer subtypes, with some treatment episodes lasting only a few weeks and others extending beyond 200 weeks. Multiple dose changes and treatment interruptions are observed across several patients, with periods of treatment interruption (white gaps) and changes in daily dose evident throughout the treatment course for most patients (Figure 1). One RCC patient did not report adverse events and remained on treatment at study end. The median time to either the first treatment break, dose reduction or discontinuation (related to adverse events) in this group was 52 days (Table 4). While the median time to onset of the first reported adverse event was 25 days overall (Table 4, Supplementary table 3).
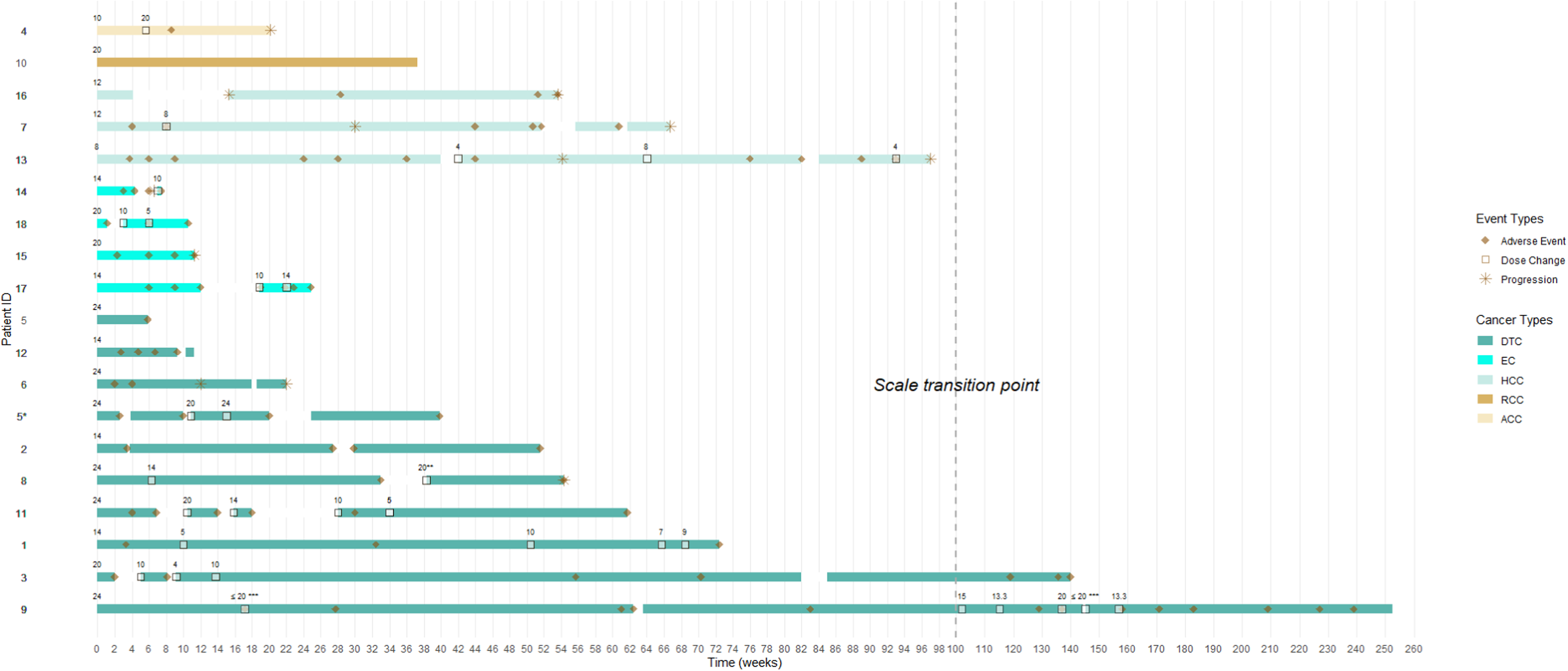
Swimmer's plot of patients treated with lenvatinib across multiple cancer types. This figure displays individual treatment duration, dosing patterns, adverse events, and treatment interruptions over time for each patient.*Patient ID 5 and 5* are the same. The patient was counted as two separate treatment episodes due to an extended treatment break.**For Patient ID 8, intermittent treatment discontinuations were observed beginning approximately 38 weeks after treatment initiation; however, the exact number and timing of subsequent breaks could not be determined from the available documentation.***For patient ID 9, treatment breaks were self-initiated as needed to manage adverse effects during certain periods of their treatment as denoted.†White gaps within each bar denote periods of treatment interruption.††Numerical values above each horizontal bar represent the lenvatinib dose (mg), reported as daily averages from the corresponding timepoint.†††The dashed vertical line indicates a change in the x-axis scale, with increments of 2 weeks before the transition and 10 weeks after.
Description of events associated with lenvatinib treatment.

†Includes a single patient (patient ID 5 and 5*) counted as 2 separate episodes for the purpose of the analysis.*One patient (patient ID: 10) was excluded from this analysis as there was no reported adverse events, dose reductions or treatment breaks for the patient and they were still on treatment beyond the follow up period.
Four treatment episodes were estimated to have discontinued treatment solely due to disease progression, while 11 treatment episodes (58%) ceased treatment for reasons related to adverse events (Figure 1, Supplementary Table 3). One patient (Patient ID: 12) was lost to follow-up, and two patients (Patient IDs: 9 and 10) continued treatment on lenvatinib beyond the follow up period (Supplementary Table 3).
Eight patients had estimable disease progression dates based on clinical documentation, of these patients, three were in the reduced starting dose group. Across the cancer types within this group, the median estimated time to disease progression was 124 days: 141 days for those that started a reduced dose and 107 days for those that started at the RLD (Supplementary Table 4).
Discussion
To the best of our knowledge, this is the first study to examine lenvatinib dosing patterns across various cancer types in an Australian public tertiary hospital. Our findings highlight a disconnect between the RLD for lenvatinib and real-world practices. Nearly half of the patient episodes (42%) initiated treatment with at an empirically reduced dose, with majority of them never being escalated to the RLD. This suggests that clinicians are aware of the poor tolerability of lenvatinib when prescribed at the RLD. However, despite empirical dose reductions, almost all treatment episodes (95%) experienced at least one lenvatinib-related adverse events, and many (58%) required treatment discontinuation due to adverse events. Together, these findings suggest that current dosing strategies may be insufficient to mitigate the significant tolerability challenges associated with lenvatinib in routine practice.
The onset and rate of adverse events reported in our study align with previous studies reporting adverse event rate of nearly 100%,2,11,30 along with a median time to the first onset of any grade adverse event of 37 days11,18,30,31 associated with lenvatinib treatment. Our data alludes that a reduced starting dose may not delay or prevent toxicity associated with lenvatinib, as the median time to the first adverse event observed in our cohort was 25 days. This suggests that empirical dose reduction of lenvatinib may not be the most optimal strategy to mitigate toxicity associated with lenvatinib.
In our study we observed an overall median 8-week RDI of 70% (IQR:55–96), which is similar to the suggested threshold reported in prior studies for favourable outcomes with lenvatinib treatment.9,10 However, given the wide variability in the 8-week RDI observed, a subset of the cohort may have been underdosed initially. This highlights a clinical dilemma: standard doses of lenvatinib may be excessive for some patients 32 while empirical dose reductions without objective guidance may compromise treatment efficacy (Supplementary Table 3).
Previous studies have highlighted intra- and interpatient variability in lenvatinib exposure and response, with an exploratory target trough concentration (Cmin) of ≥51.5 ng/mL proposed.5,23,33–36 Given the association between exposure and response, TDM a form of precision dosing which involves measuring plasma drug levels, offers a potential approach to guide the dosing of lenvatinib objectively.13,23–25 Furthermore, lenvatinib may be a suitable candidate for TDM due to its lack of active metabolites and the availability of rapid quantification methods requiring minimal sample volume. 23 However, no pharmacokinetic measurements were performed in this study; therefore, no conclusions regarding exposure–response relationships can be drawn. The role of TDM in this context should be considered hypothesis-generating and requires further prospective evaluation.
Oncology pharmacists may be well positioned to contribute to the development, evaluation and implementation of TDM-guided approaches within multidisciplinary teams. Oncology pharmacists routinely assess treatment tolerability, review adherence, manage drug interactions, and support dose modification of cancer drugs alongside oncologists, all of which are core functions that directly align with precision dosing. 27 A recent qualitative study reports that pharmacists are willing to champion TDM initiatives in oncology as they have the adequate pharmacokinetics expertise and knowledge required to drive this. 26 Successful implementation, however, requires a coordinated, systems-level framework, including standardised protocols, high quality evidence, prospective studies, multidisciplinary engagement, timely access to assays, and integration with existing clinical workflows. 26 Such infrastructure will be essential if TDM-guided dosing is to be adopted more broadly in oncology practice.
This study has limitations inherent to its design. First, the small sample size substantially limits the generalisability of the findings. The ability to detect meaningful differences between subgroups is restricted and thus the results should be interpreted as exploratory and reflective of local practice. Second, the retrospective design introduces risks of selection and information bias. Data on adverse events, disease progression, dosing regimen and reasons for dose modification were dependent on clinical documentation, which may vary in consistency. This limited the scope and depth of analyses that could be performed. Additionally, relevant clinical variables, such as performance status and biomarkers (e.g., thyroglobulin, alpha-fetoprotein), were not systematically recorded and therefore could not be included in the analysis. The use of medication records to infer comorbidities may also have introduced inaccuracies, particularly in cases where medical histories were incomplete, or medications were prescribed for off-label indications. Third, as this was a single-centre study, the findings may reflect local prescribing practices, clinician preferences, and patient demographics, limiting the external validity of the findings. Fourth, adherence to oral lenvatinib was assumed to be 100% unless otherwise documented. In Australia, lenvatinib is subsidised under the Pharmaceutical Benefits Scheme for multiple cancer indications, which may reduce financial barriers and support medication adherence.37–40 Nevertheless, this assumption may not fully reflect real-world patient behaviour and could result in overestimation of true drug exposure and relative dose intensity. Fifth, the inclusion of multiple cancer types with differing dosing strategies and toxicity profiles introduces clinical heterogeneity. However, this reflects the real-world use of lenvatinib across indications and was intentional, as the aim of this study was to evaluate overall dosing practices rather than perform tumour-specific comparisons. As such, the findings should be interpreted as descriptive across indications rather than disease specific. Finally, adverse events were identified from clinical documentation and were not graded using standardised criteria such as the CTCAE. This introduces subjectivity and potential variability in reporting and limits the ability to compare toxicity severity across patients. Despite this, the high rate of adverse events and dose modifications observed aligns with international data and highlights the real-world challenges with lenvatinib treatment.
This study highlights substantial variability in lenvatinib dosing in routine clinical practice at a single centre. Despite an upfront dose reduction, some patients still experience adverse events and treatment interruptions. While these findings highlight variability in response to treatment and real-world dosing, they should be interpreted cautiously given the small cohort size. Opportunities exist to improve dose selection for lenvatinib, though further studies in larger populations are required. Oncology pharmacists may be well positioned to contribute to the development, evaluation, and implementation of TDM-guided approaches within multidisciplinary teams. Future research should focus on defining clinically relevant exposure targets and establishing the systems-level infrastructure needed to support TDM-guided dosing of lenvatinib.
Supplemental Material
sj-pdf-1-opp-10.1177_10781552261459659 - Supplemental material for Lenvatinib use across multiple cancer types: A single-centre evaluation of dosing patterns, tolerability and opportunities for dose optimisation
Supplemental material, sj-pdf-1-opp-10.1177_10781552261459659 for Lenvatinib use across multiple cancer types: A single-centre evaluation of dosing patterns, tolerability and opportunities for dose optimisation by Jin Quan Eugene Tan, Natansh D Modi, Ramin Hassankhani, Helen Martin, Madele van Dyk, Shane Spencer, Benedetta C Sallustio and Ganessan Kichenadasse in Journal of Oncology Pharmacy Practice
Footnotes
CRediT author statement
Funding
This work was supported by the Ryan Hodges Fund, Flinders Foundation.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.