
Abstract
Chronic kidney disease (CKD) is imperceptible and with a complex pathogenesis involving oxidative stress and inflammatory responses. The phosphatidylinositol 3 kinases/protein kinase B (PI3K/AKT) signaling pathway plays a crucial role in this process. Previous studies indicated that chemokine ligand 25 (CCL25) and its receptor CCR9 participate in regulating the PI3K/AKT pathway. However, it remains unclear whether CCL25/CCR9 influences CKD by modulating oxidative stress and inflammation via the PI3K/AKT pathway. We aimed to clarify the roles of CCL25/CCR9 in CKD progression through PI3K/AKT activation and its upstream regulatory factors. In this study, an adenine-induced CKD rat model and an indoxyl sulfate-induced CKD cell model were constructed. Bioinformatics method based on two online websites (JASPAR and Cistrome DB) was conducted to predict the upstream of CCL25/CCR9. Anti-CCL25 and PI3K/AKT activator 740Y-P were added, and sh-IRF1 was transfected into the HK-2 cells for the mechanism exploration. Renal function markers (blood urea nitrogen, creatinine, urine protein/creatinine ratio), renal pathology, oxidative stress markers (reactive oxygen species, malondialdehyde, superoxide dismutase), inflammatory cytokines (Interleukin (IL)-1β, IL-6, Tumor Necrosis Factor (TNF)-α), and the expression of CCL25, CCR9, PI3K/AKT pathway-related proteins, and the transcription factor interferon regulatory factor 1 (IRF1) were measured. Chromatin immunoprecipitation PCR (ChIP-PCR) was used to confirm IRF1 binding to the CCL25 promoter. The results indicated that CCL25 and CCR9 were upregulated in CKD rat and in vitro cell model. Enhanced oxidative stress and inflammatory response, followed by renal fibrosis and renal dysfunction, were observed in CKD rats. Anti-CCL25 addition decreased the PI3K/AKT markers, oxidative stress markers, and inflammatory responses, whereas 740Y-P addition reversed the above alteration. IRF1 was confirmed to be an important transcription factor for CCL25 in bioinformatics analysis. IRF1 was increased in CKD models and binding with CCL25 in ChIP-PCR detection. sh-IRF1 transfection decreased the CCL25 and CCR9 expression. In conclusion, IRF1-regulated CCL25/CCR9 was demonstrated to promote CKD progression through PI3K/AKT-mediated oxidative stress and inflammatory response.
Keywords
Introduction
Kidney diseases are a pivotal public health issue, commonly including acute kidney injury and chronic kidney disease (CKD). Among which, CKD is defined as renal function loss or decreased glomerular filtration rate lasting for at least 3 months, which is irreversible and eventually leads to end-stage renal disease (ESRD) (Lameire et al., 2021). CKD affects 697.5 million people worldwide, and its prevalence rate is constantly rising (Siracusa et al., 2024). Once CKD progresses to ESRD, the only effective strategy is renal replacement therapy, which includes dialysis or kidney transplantation (Bakinowska et al., 2024). However, these treatments are expensive and may induce high morbidity and mortality (Liu et al., 2024). Moreover, the underlying pathogenesis mechanisms of CKD remain insufficient. Hence, it is urgent to explore novel therapeutic targets to improve the prevention and treatment of CKD.
CKD refers to a process of chronic changes in the kidneys, with clinical characteristics such as chronic inflammation, renal fibrosis, glomerular sclerosis, and sparse blood vessels (Hodgin et al., 2024). Its pathogenesis is related to various alterations, including metabolic and hormonal dysregulation, as well as low-grade systemic inflammation (Bakinowska et al., 2024). Oxidative stress and inflammatory regulation alteration have recently been reported to participate the CKD progression (Kishi et al., 2024). For example, Li et al. indicated that leukocyte telomere length affects the prognosis and renal function through regulation of oxidative stress in CKD condition (Yi et al., 2025). In the processes of regulation of oxidative stress and inflammatory response, the important roles of phosphatidylinositol 3 kinases (PI3K)/protein kinase B (AKT) signaling pathway are emphasized (Wang et al., 2023; Wang et al., 2024). The inhibition of PI3K/AKT signaling pathway is reported to participate in various biological process, including inflammation, oxidative stress, epithelial–mesenchymal transformation, and autophagy, to ameliorate kidney damage (Wang et al., 2024). Hence, we hypothesized that the PI3K/AKT pathway can influence the development of CKD by regulating oxidative stress and inflammation response.
The mechanism by which targeting the PI3K/AKT pathway affects CKD has not been fully elucidated. The interaction between chemokines and their receptors has been proven to be an important biomarker and therapeutic target for kidney diseases (Chang and Chen, 2020). This interaction participates in the progression and treatment of CKD by influencing immune cells, inflammatory response, oxidative stress, and renal function (Chang et al., 2024; Yoo et al., 2024). Chemokine ligand 25 (CCL25) and its sole receptor CCR9 have been demonstrated to participate in the PI3K/AKT pathway in various diseases, including cancer, cardiovascular diseases, and inflammatory disorders (Hao et al., 2024; Hara et al., 2024; Wu et al., 2025; Yu et al., 2023). Additionally, CCL25 is upregulated in kidney diseases, including IgA nephropathy and clear cell renal cell carcinoma (Liu et al., 2023; Zheng et al., 2024), indicating the crucial regulatory significance of CCL25 in kidney diseases. However, it is not yet clear whether CCL25/CCR9 can also participate in the PI3K/AKT pathway-mediated oxidative stress and inflammatory response in kidney disease.
In summary, although recent studies have highlighted the pivotal role of PI3K/AKT pathway in CKD and the involvement of CCL25/CCR9 in kidney diseases, the precise upstream regulator of CCL25 and the functional link between CCL25/CCR9 and the PI3K/AKT pathway in the context of CKD remain largely unexplored. Therefore, the novelty of this study lies in: (1) mechanistically demonstrating that the CCL25/CCR9 axis exacerbates CKD progression specifically by activating the PI3K/AKT pathway, thereby amplifying oxidative stress and inflammatory damage; (2) identifying and validating interferon regulatory factor 1 (IRF1) as a key upstream transcriptional regulator of CCL25 in CKD, a finding not reported previously; and (3) integrating upstream transcriptional regulation (IRF1), chemokine signaling (CCL25/CCR9), and a core kinase pathway (PI3K/AKT) into a coherent pathogenic axis in CKD models. Specifically, an adenine-induced rat CKD model and an indoxyl sulfate (IS)-induced in vitro CKD model were constructed to verify the above hypothesis. This study uncovers the potential regulatory mechanism based on IRF1/CCL25/CCR9/PI3K/AKT axis and provides novel insight into the prevention and treatment of CKD.
Materials and Methods
Animal experiments
Twelve SD rats (150–200 g) were acquired from Beijing Huafukang Biotechnology Co., Ltd. (Beijing, China). All the rats were cultured under a 12-h light/dark cycle at 20–25°C with the humidity of 50–55%. After adoption for 5 days under conditions of free access to water and feed, all the rats were randomly grouped into two groups (N = 6): control and CKD model. The rats in control groups received 1% sodium carboxymethyl cellulose (C8621, Thermo) and standard diet. The rats in the CKD model groups received adenine (A8330, Solarbio) supplementation in the diet (0.75% w/w adenine in the standard diet) to induce CKD model as reported previously (Ghelani et al., 2019).
On the 21st day, all the rats were kept alone in metabolic cages and allowed to adapt for another 2 days. On the 24th day, 24-h feces and urine samples were collected. Then the rats were anesthetized using Zoletil (0.5 g/100 g, Yangzhou University Animal Hospital), blood and kidney tissues were collected. Finally, the rats were euthanized by carbon dioxide asphyxiation method. During the period of animal modeling culture, the changes in animal weight were recorded every day.
All the animal experiments were complied with ARRIVE guidelines and were approved by the Experimental animal Ethics Committee of Yangzhou University (NO. 202509057).
Cell experiments
The human renal tubular epithelial cells HK-2 (CL-0109, Wuhan Procell) were cultured in DMEM/F12 medium (11320033, Thermo) containing 10% fetal bovine serum (34080619, Ephraim) and 1% penicillin/streptomycin solution at 37°C, 5% CO2, and 100% humidity. When the cell density was approximately 90%, regular passages were performed. The 4th generation cells were used for subsequent experiments.
The cell experiments consisted of two parts. In the first, the HK-2 cells were treated with 250 μM IS for 24 h to establish an in vitro CKD model. Then, the cells were cultivated with 10 μL of 200 ng/mL anti-CCL25 for 48 h, or were treated with 20 μM PI3K pathway activator (740Y-P) for 24 h. In the second part, the sh-NC and sh-IRF1 lentivirus were transfected into both the HK-2 cells and IS-treated HK-2 cells. The sh-IRF1 sequence was designed in VectorBee (https://www.vectorbuilder.cn/) and shown as: TGGCTAGAGATGCAGATTAAT.
Blood urea nitrogen and serum creatinine detection
The whole blood samples of SD rats were extracted through cardiac puncture and collected in 15-mL centrifuge tubes. Then, the blood samples were centrifuged at 1000 g for 15 min, and the upper layer serum was collected for detection of blood urea nitrogen (BUN) and creatinine (CR) using an automatic biochemical analyzer (Indiko, Thermo). The used kit was BUN (BC5655, Solarbio) and CR (BC4915, Solarbio).
Ratio of urine protein to urine CR (RuP/uCr) detection
The urine samples of each rat within 24 h before their sacrifice were collected. The kit was adopted to detect the contents of urine protein and urine CR, and RuP/uCr of 24 h for the rats was calculated.
Hematoxylin–eosin staining
Hematoxylin–eosin (H&E) kit (C0105S, Beyotime) was applied to evaluate the pathological changes of rat kidneys. The tissue was fixed in 4% methanol, dehydrated using ethyl alcohol, paraffin-embedded, and sliced at 4–7 μm. The section was stained with hematoxylin for 5 min and with eosin for 1–2 min, and dehydrated with xylene and ethanol.
ELISA detection
The levels of inflammatory cytokines (IL-1β, IL-6, and TNF-α) in the serum of the animal and the HK-2 cells were measured using the corresponding ELISA-related IL-1β assay kit (ml037361, Mlbio; ml028592, Mlbio), IL-6 assay kit (ml064292, Mlbio; SEKH-0013, Solarbio), and TNF-α assay kit (ml002859, Mlbio; ml064303, Mlbio). The levels of inflammatory cytokines were evaluated at 450 nm absorbance.
Reactive oxygen species detection
Reactive oxygen species (ROS) in the kidney tissues were detected using Dihydroethidium/Ethidium (DHE/ET) fluorescence quantitative method. The fresh tissue sample was washed using phosphate-buffered saline (PBS; C0221A, Beyotime), supplemented with 1 mL of homogenization buffer A for every 50 mg of tissue to conduct the tissue homogenization. After centrifugation at 100 g for 3 min at 4°C, the supernatant was added with 2 μL of DHE probe to culture for 15–30 min at 37°C in the dark. The fluorescence intensity was measured using a fluorescence microplate reader (SpectraMax M4, MolecularDevices) at an excitation wavelength of 488–535 nm and an emission wavelength of 610 nm.
ROS in cells were detected using flow cytometry. Cell suspensions were collected by centrifuging at 250g for 5 min. Then, the precipitate was collected for resuspend using PBS and centrifuging at 250g for another 5 min. DCFH-DA probes were added to the cells to incubate for 20 min at 37°C. The ROS levels were detected using flow cytometry (CytoFLEX s, Beckman) at an excitation wavelength of 488 nm and an emission wavelength of 525 ± 20 nm for detection.
Malondialdehyde and superoxide dismutase detection
For the tissue preprocessing, the fresh tissue sample was washed using PBS, supplemented with 1 mL of homogenization buffer A for every 50 mg of tissue to conduct the tissue homogenization. After centrifuging at 100g for 3 min at 4°C, the supernatant was collected. For the cell preprocessing, the cell culture supernatant was discarded, and 0.5 mL of PBS was added to the cell precipitate. The cells were placed in an ice water bathe with the power of 300 W. After four rounds of sonication (3–5 s per pulse with 30 s intervals), the cells were lysed into a suspension.
Malondialdehyde (MDA) and superoxide dismutase (SOD) levels in renal tissue and HK-2 cells were detected using the corresponding MDA kit (A003-1-2, Nanjing Jiancheng) and SOD kit (A001-3, Nanjing Jiancheng), respectively.
Cell viability detection
Cell viability in each group was detected using the CCK-8 method. Briefly, 10 μL of CCK-8 reaction solution (C0037, Beyotime) was supplemented into 96-well plates with cell suspension (100 μL, 2,000 cells/well). After 2 h of culture, the cell viability was measured using a microplate reader.
qRT-PCR detection
Total RNA was extracted from kidney tissue and HK-2 cells using a TRIzol reagent (15596018, Invitrogen). Gene expression was computed using the 2–ΔΔCt method with GAPDH as the internal control. The primers used in this study are listed in Table 1.
Primers Used in This Study

CCL25, chemokine ligand 25.
Western blotting
The proteins (CCL25, CCR9, PRDM1, and IRF1) and PI3K/AKT pathway proteins (PI3K, p-PI3K, AKT, and p-AKT) were detected using western blotting method. The protein was isolated from kidney tissue or HK-2 cells using radioimmuno-precipitation assay lysis buffer (P0013B, Beyotime), and then the protein concentration was quantified using a bicinchoninic acid kit (1610374, Beyotime). GAPDH was considered as the internal control. Primary antibodies used in the study were shown as follows: anti-CCL25 (1:1000; PA5-116019, Thermo), anti-CCR9 (1:1000; PA5-95131, Thermo), anti-PRDM1 (0.5 μg/mL; PA5-79861, Thermo), anti-IRF1 (0.5 μg/mL; PA5-147374, Thermo), anti-PI3K (1:1000; PA5-29220, Thermo), anti-p-PI3K (1:1000; PA5-105114, Thermo), anti-AKT (1:1000; 44-609 G, Thermo), anti-p-AKT (1:500; PA5-36780, Thermo), and anti-GAPDH (PA1-987, Thermo).
Chromatin immunoprecipitation polymerase chain reaction detection
Chromatin immunoprecipitation polymerase chain reaction (ChIP-PCR) was used to determine IRF1 binding in CCL25. An enzymatic chromatin IP kit (WLA106, WanleiBio) was used to extract chromatin and DNA according to the manufacturer’s protocol. Anti-IRF1 antibody, polymerase Ⅱ (positive control), and IgG (negative control) were used for ChIP.
Statistical analysis
GraphPad 10.0 software was employed for statistical analyses and plot visualization. All data are expressed as mean ± standard deviation. One-way analysis of variance with Tukey’s post hoc test was used for comparing the differences among multiple groups. Differences between groups were considered statistically significant if P < 0.05.
Results
Upregulated CCL25 and CCR9 in CKD rats
To investigate the expression levels of CCL25 and CCR9 in CKD, a CKD rat mouse was established by adenine supplementation. During the growth process, the body weight of rats in CKD and control groups was gradually increased, with no significant differences (Fig. 1A). The biochemical indexes BUN, CR, and RuP/uCr in CKD rats were significantly upregulated when compared with control group (Fig. 1B). H&E staining indicated that the renal structure in the CKD group was disordered when compared with the control group, manifesting as reduced glomeruli number and shrunken glomeruli, significantly expanded cyst cavities and the renal tubules, ruptured lumen, reduced interstitial blood vessels, increased interstitial fibrosis, and present interstitial inflammation (Fig. 1C). These findings indicated injury and fibrosis of kidney in CKD rat. The expression levels of CCL25 and CCR9 were both increased in the CKD rat than that in control rat (Fig. 1D).
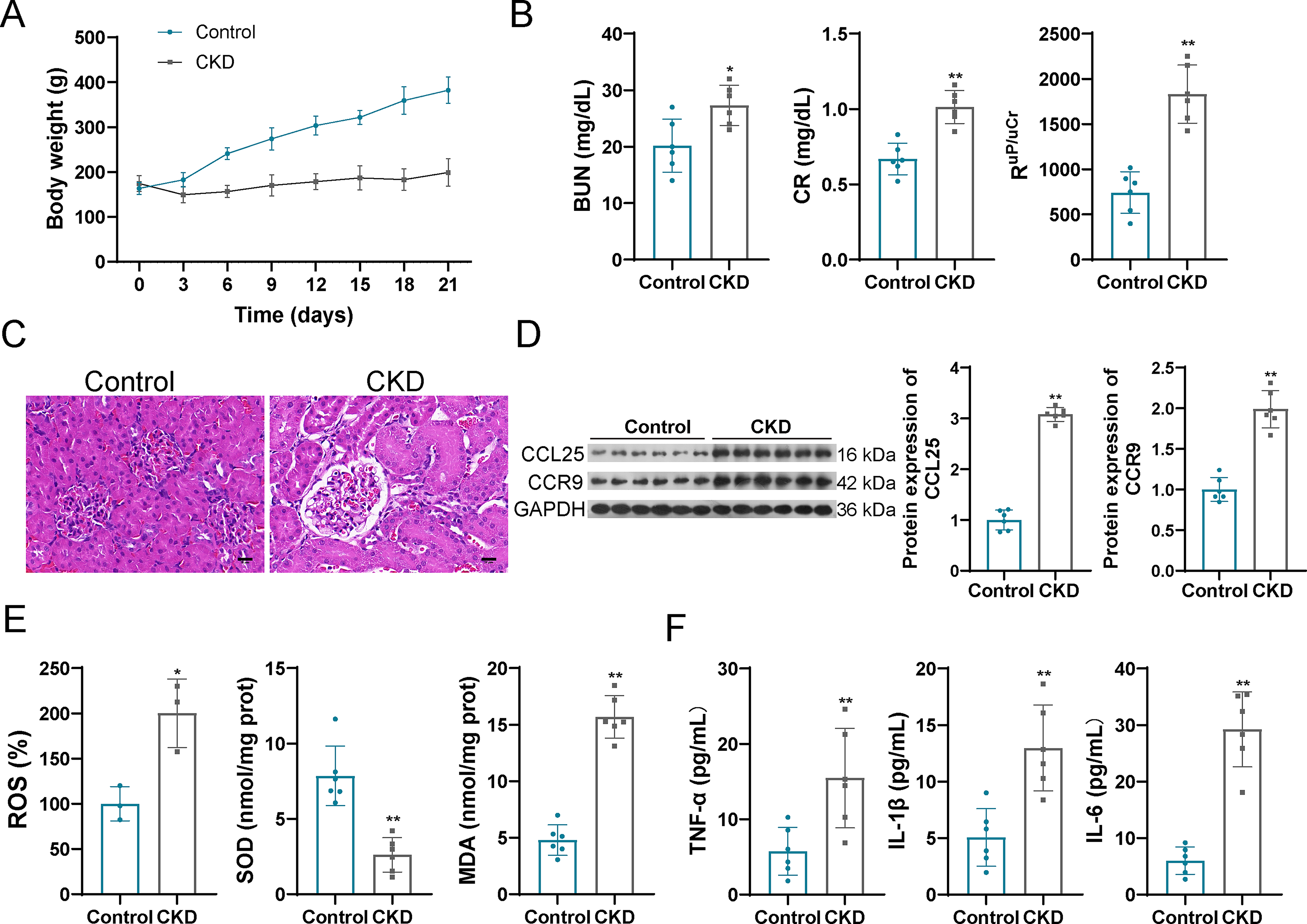
Upregulated CCL25 and CCR9, increased oxidative stress, and inflammatory response in CKD rat.
Increased oxidative stress and inflammatory response in CKD rat
Increased ROS and MDA levels, as well as decreased SOD levels in CKD samples were observed, indicating activation of oxidative stress in CKD rats (Fig. 1E). Simultaneously, the pro-inflammatory cytokines TNF-α, IL-6, and IL-1β were selected for the evaluation of inflammatory intensity because they are established central mediators in the pathogenesis of CKD (Maia et al., 2024). As expected, levels of IL-1β, IL-6, and TNF-α were also significantly elevated in CKD rats compared with controls (Fig. 1F), consistent with an enhanced inflammatory response.
CCL25/CCR9 promotes oxidative stress and inflammatory responses by activating the PI3K/AKT pathway in cells
CCL25/CCR9 interaction has been demonstrated to be involved in the PI3K/AKT pathway-related biological processes (Chai et al., 2022). To verify the mechanism of CCL25/CCR9-activated PI3K/AKT pathway in CKD, the normal HK-2 cell line was cultured with IS to construct a CKD cell model. Then, a CCL25 inhibitor (anti-CCL25) was added to the CKD cells. Like the results in the CKD rat model, increased CCL25 and CCR9 levels were also observed in the CKD cell model (Fig. 2A). Upon addition of anti-CCL25, the levels of CCL25 and CCR9 were significantly decreased (Fig. 2A). In the IS-treated HK-2 cell model, PI3K/AKT pathway proteins were upregulated, showing as increased p-PI3K/PI3K and p-AKT/AKT in CKD cell model compared with that in control cells (Fig. 2A); however, after addition of anti-CCL25, the increased PI3K/PI3K and p-AKT/AKT in model cells were further decreased (Fig. 2A). Moreover, the cell viability indicated a similar trend under the anti-CCL25 addition, that is, decreased cell viability in IS-treated cell model was further increased by anti-CCL25 addition (Fig. 2B). It is worth noting that, activation of oxidative stress and inflammatory response in the CKD cell model were also observed, showing as increased ROS, MDA, IL-1β, IL-6, and TNF-α levels, as well as decreased SOD level in the IS-treated HK-2 cells (Fig. 2C–E). However, the above alterations were also reversed by an anti-CCL25 addition (Fig. 2C–E). These results indicated that activation of PI3K/AKT pathway, oxidative stress, and inflammatory responses was regulated by CCL25.
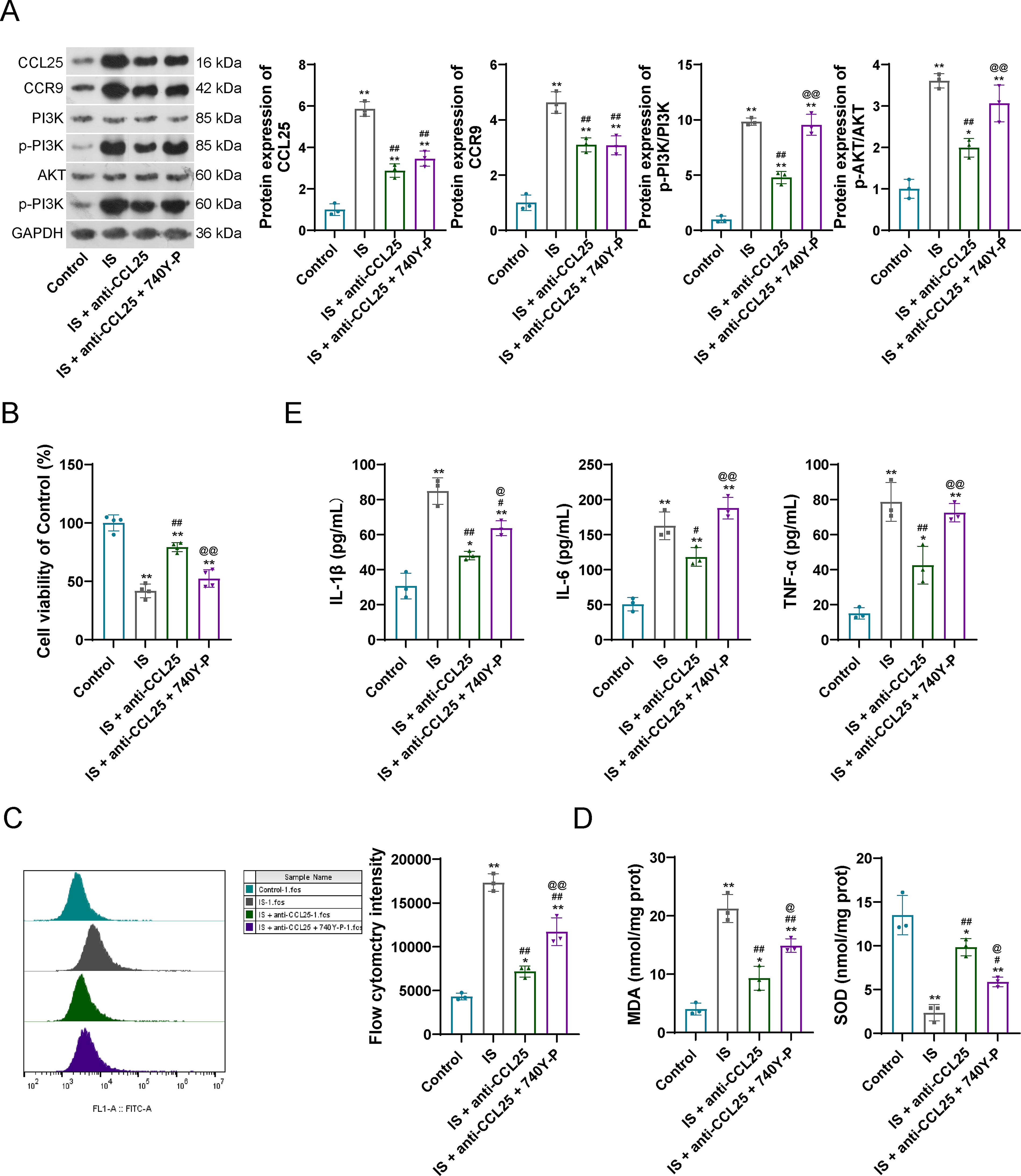
CCL25/CCR9 promotes oxidative stress and inflammatory responses by activating the PI3K/AKT pathway in cells.
Subsequently, a PI3K activator (740Y-P) was added into the CKD cells containing anti-CCL25 to verify the regulatory effects of PI3K/AKT pathway on the downstream oxidative stress and inflammatory responses. As expected, coincubation of anti-CCL25 and 740Y-P exhibited little influence on the CCL25 and CCR9 expression, but significantly upregulated the p-PI3K/PI3K and p-AKT/AKT levels (Fig. 2A). Moreover, coincubation of anti-CCL25 and 740Y-P decreased the cell viability when compared with only anti-CCL25 treated model cells (Fig. 2B). Also, coincubation of anti-CCL25 and 740Y-P reactivated the oxidative stress and inflammatory responses (Fig. 2C–E).
The above findings fully proved that CCL25/CCR9 could promote oxidative stress and inflammatory responses by activating the PI3K/AKT pathway in IS-treated CKD cell models.
Transcription factor IRF1 regulates the CCL25 expression
To search for the upstream regulatory factors of CCL25, a bioinformatics analysis was carried out to predict the effective upstream transcription factors (TFs) of CCL25 based on two websites: JASPAR and Cistrome DB. A total of 51 TFs and 9 TFs were confirmed in JASPAR and Cistrome DB, respectively. After intersecting the TFs resulted from the two sources, only IRF1 and PRDM1 were confirmed to be the common TFs targeted to CCL25 (Fig. 3A). The binding sites of the two TFs that targeted CCL25 are visualized in Figure 3B. The expression levels of IRF1 were significantly increased in the CKD rat model, while PRDM1 showed no significant changes in CKD condition when compared with control rat (Fig. 3C and D). Hence, IRF1 was served to be an important regulatory factor to CCL25, and its expression levels were also increased in IS-treated CKD cell models (Fig. 3E).
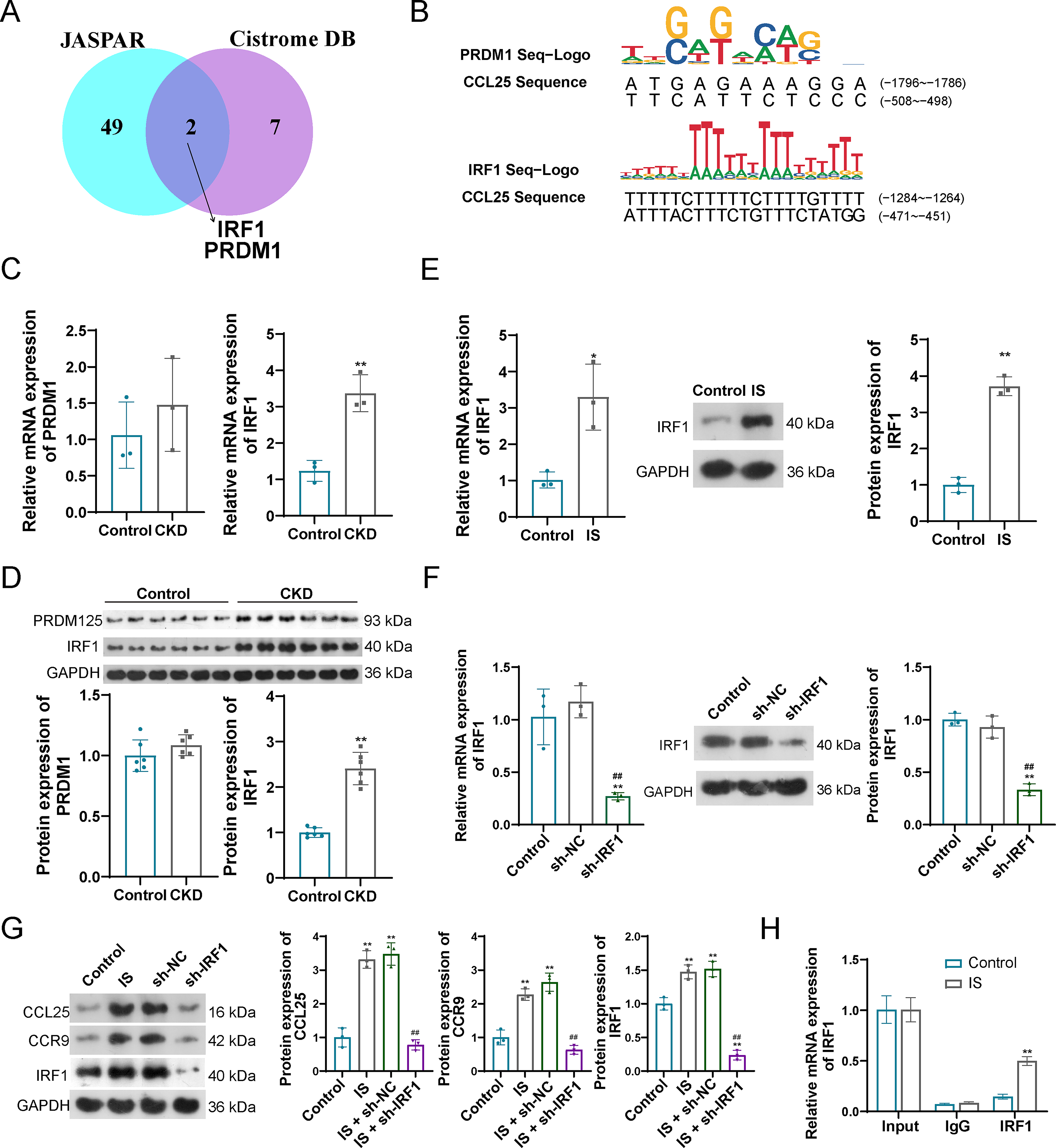
Transcription factor (TF) IRF1 regulates the CCL25 expression.
To verify the regulatory fact of IRF1 on CCL25/CCR9, IRF1 was effectively knockdown by the transfection of sh-IRF1 in HK-2 cells (Fig. 3F). When these IRF1-knockdown cells were stimulated with IS, the increased expression levels of CCL25 and CCR9 (observed in the IS-treated CKD models) were significantly attenuated. Notebaly, the reduction in CCL25 and CCR9 expressions paralleled the decrease in IRF1 levels (Fig. 3G). Moreover, ChIP-PCR indicated that the combination of IRF1 with CCL25 promoter, and IS treatment promotes the binding of IRF1 to CCL25 (Fig. 3H).
Discussion
CKD poses a threat to human health and survival, encouraging us to identify pathogenic mechanisms and potential therapeutic targets. In the present results, IRF1-regulated CCL25/CCR9 was demonstrated to promote CKD progression through PI3K/AKT-mediated oxidative stress and inflammatory response (Fig. 4).
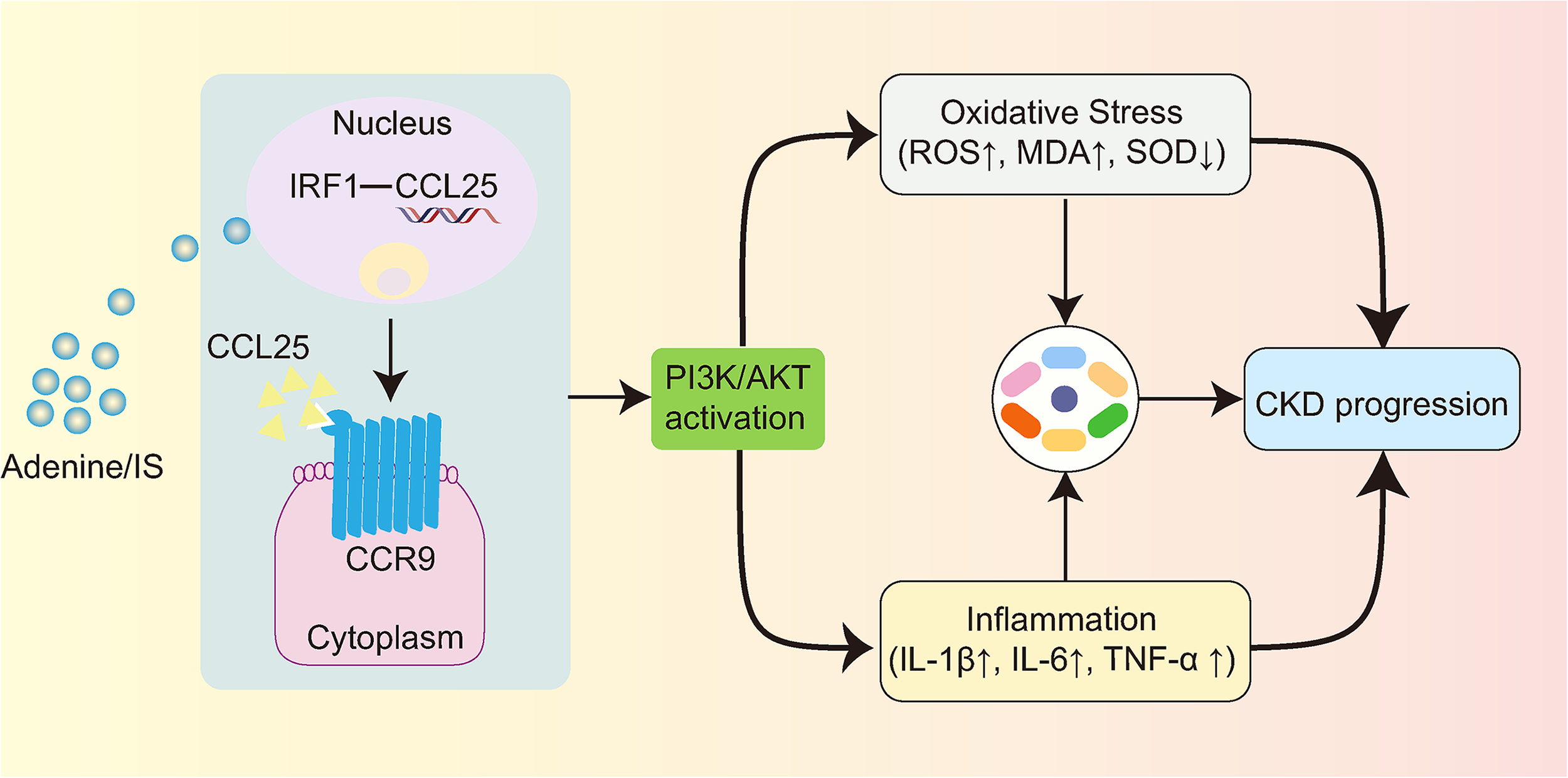
IRF1-regulated CCL25/CCR9 promotes CKD progression through PI3K/AKT-mediated oxidative stress and inflammatory response. CCL25, chemokine ligand 25; CKD, chronic kidney disease; PI3K/AKT, phosphatidylinositol 3 kinases/protein kinase B.
In this study, an adenine-induced rat mode was selected for the investigation because it is a well-established and reproducible model that recapitulates key features of human CKD (Diwan et al., 2018); and an IS-treated HK-2 cell model was employed because IS is a prototypical and clinically relevant uremic toxin that accumulates in CKD and directly drives tubular injury by inducing oxidative stress and inflammation (Düsing et al., 2025)—key processes under investigation. Moreover, both adenine and IS are involved in toxin-mediated tubular injury, thereby ensuring mechanistic consistency between our in vivo and in vitro findings. Renal dysfunction, renal fibrosis, chronic inflammation, and oxidative stress are the main pathological alterations and features of CKD (Hodgin et al., 2024; Kishi et al., 2024), which were also verified in our rat and in vitro cell CKD models. Increased BUN, CR, RuP/uCr, oxidative stress markers (ROS and MDA), and inflammatory factors (IL-1β, IL-6, and TNF-α), as well as decreased oxidative stress marker SOD, proved this.
The regulatory mechanisms of chemokine–receptor complex have been emphasized in various morbid states, including renal fibrosis and renal dysfunction (He et al., 2023; von Vietinghoff and Kurts, 2021). For example, C-C motif chemokine ligand 2 (CCL2), together with its major receptor CCR2, are activated in kidney diseases, and mediate and induce renal fibrosis through recruiting monocytes (He et al., 2023). CCL25/CCR9 interaction has also been proven to involve fibrosis-related diseases, such as myocardial fibrosis and pulmonary fibrosis (Morikawa et al., 2021; Nakamoto, 2016; Wu et al., 2025). However, CCL25/CCR9 interaction has not been fully clarified in renal fibrosis. In our investigation, CCL25 and CCR9 were upregulated both in the rat and in vitro cell CKD models; and the inhibition of CCL25 in vitro prevented the intensification of inflammatory responses and oxidative stress, thereby promote the cell viability. These findings indirectly indicated the involvement of CCL25/CCR9 in CKD progression.
Moreover, through the addition of a PI3K activator (740Y-P) into the in vitro cells, the CCL25/CCR9-induced oxidative stress and inflammatory responses were demonstrated to be mediated by a classical PI3K/AKT pathway. PI3K/AKT plays a pivotal role in the regulation of cell function, which includes cell proliferation, differentiation, and metabolism (Liu et al., 2024). PI3K is an enzyme involved in neural signal transduction and is reported to be activated by various receptors such as tyrosine kinases, G protein-coupled cytokines, and Ras-related GDPase receptors (Yu et al., 2022). In our investigation, PI3K was demonstrated to be activated by CCL25/CCR9 interaction. AKT is a downstream target of PI3K and is also significant in regulating various cellular functions (Sementino et al., 2024). In our in vitro cell CKD models, high expression levels of PI3K and AKT demonstrated the activation of PI3K/AKT cascade reaction in CKD; and inhibition of CCL25 inhibited the PI3K/AKT signaling pathway. PI3K/AKT pathway and its cascade reaction regulate the renal fibrosis and renal functions in various studies through lipid metabolism, oxidative stress, and inflammatory responses (Wang et al., 2024; Hu et al., 2025; Xia et al., 2025; Yang et al., 2019), which is also coincidence with our results.
IRF1 is a mammalian TF and is highly upregulated in CKD (Li et al., 2020). The overexpression of IRF1 promotes the risk of renal fibrosis through various mechanisms, such as downregulation of Klotho (Li et al., 2020) and TWEAK/Fn14-mediated ECM (Gu et al., 2023). These evidences are similar to our research that IRF1 is a risk factor in CKD. We also proposed that IRF1 participated in the mechanism of renal fibrosis through a new cytokine–receptor interaction (CCL25/CCR9). The binding ability of IRF1 with CCL25 was verified in a ChIP-PCR detection, and knockdown of IRF1 suggested decreased expression of both CCL25 and CCR9. These results indicated that IRF1 might be a risk indicator and a therapeutic target in CKD.
Our study firstly demonstrated the involvement of CCL25/CCR9 interaction in the PI3K/AKT-mediated inflammatory and oxidative stress-related pathological process in CKD, and novelly clarified its upstream TF IRF1. However, there are still some limitations. First, the specific functions and regulatory mechanisms of CCL25/CCR9 and its upstream IRF1 on renal fibrosis are less to be mentioned, whether in this study or in previous publications. Second, whether IRF1 and CCL25/CCR9 can be applied to the prognostic biomarkers or therapy targets in CKD remains unknown. Finally, our studies were mainly focused on the mechanism based on PI3K/AKT-mediated inflammatory and oxidative stress, whether CCL25/CCR9 regulates renal fibrosis through other pathways and processes, such as NF-κB, NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, and JAK-STAT signaling, are still unknown.
Conclusion
Chemokine–receptor interaction (CCL25/CCR9) is demonstrated to involve the renal fibrosis in CKD progression, with its upstream of IRF1 and downstream mechanism based on PI3K/AKT-mediated oxidative stress and inflammatory response. The results proposed the potential of IRF1 and CCL25/CCR9 as the prognostic biomarkers and therapeutic targets in CKD, providing novel insights into the CKD management and prevention.
Submission Declaration and Verification
The work presented in this study has not been previously published, is not being considered for publication elsewhere, has been approved for publication by all authors and the relevant authorities, and will not be published in any other form without consent.
Ethical Approval
All the animal experiments were complied with ARRIVE guidelines and were approved by the Experimental animal Ethics Committee of Yangzhou University (NO. 202509057).
Authors’ Contributions
S.Y.: Conceptualization, data curation, and writing—original draft. Y.C.: Data curation, formal analysis, and writing—original draft. H.L.C.: Formal analysis and validation. C.J.: Investigation and validation. S.L.: Visualization and writing—review and editing. L.C.: Conceptualization, funding acquisition, and writing—review and editing. All authors reviewed and approved the final version.
Footnotes
Author Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Information
This research was supported by Wenzhou Basic Public Welfare Research Project (No. Y2023771).
Data Availability Statement
Data will be made available on request.