
Abstract
Purpose:
Extend Phase 2A study to evaluate additional concentrations of H-1337 and dosing frequencies, and to compare them with a positive control.
Design:
Phase 2B, randomized, double-masked, active-controlled, dose–response study of 28 days of four treatments: H-1337 0.6% b.i.d., 1.0% b.i.d., or 1.0% q.d. (1.0% in the morning with H-1337 vehicle in the evening), and timolol maleate 0.5% b.i.d.
Participants:
Two hundred one subjects with open-angle glaucoma or ocular hypertension at eight private practice sites in the United States.
Methods:
Diurnal intraocular pressure (IOP) over 28 days of dosing.
Main Outcome Measures:
Non-inferiority to timolol in change from baseline in IOP at Day 1 and Day 28.
Results:
Mean reduction in IOP was 4–7 mmHg for the H-1337 groups and 5–8 mmHg for the timolol group. Non-inferiority to timolol for H-1337 1.0% b.i.d. [upper limit of 95% confidence interval (CI) strictly lower than 1.5 mmHg] was met at 6/9 time points (Day 1: h 8 and 12; Day 28: h 2, 4, 8, and 12). Similar comparative efficacy was seen for the other H-1337 treatment groups. The most common adverse event observed was hyperemia, reported in 54.0% (27/50) for H-1337 1.0% q.d., 33.3% (17/51) for H-1337 0.6% b.i.d., 41.2% (21/51) for H-1337 1.0% b.i.d., and 8.2% (4/49) for timolol.
Conclusion:
H-1337 in doses of 0.6% b.i.d., 1.0% q.d., and 1.0% b.i.d. had ocular hypotensive efficacy in the range of timolol, although not within the strict Phase 3 non-inferiority margins, which would have required a larger sample size.
Introduction
The primary method for the treatment of open-angle glaucoma is the lowering of intraocular pressure (IOP) by either medical, surgical, or laser therapy.1,2 Among the newer pharmacological agents for lowering IOP are the rho-kinase inhibitors (RKIs).3,4 The RKIs netarsudil and ripasudil are approved in selected countries as both monotherapy and fixed-dose combinations.5–11 Each of these agents has advantages and disadvantages. 4
In an effort to provide additional agents in this class, H-1337, an RKI, is being evaluated as a topical ocular hypotensive therapy. Administered topically to the eye, H-1337 demonstrated ocular hypotensive activity in normotensive rabbits and monkeys of 7.3 mmHg and 4.3 mmHg, respectively. 12
Hartman et al. conducted a Phase 1/2 double-masked, dose–response, placebo-controlled study of the ocular hypotensive efficacy of H-1337 ophthalmic solution in subjects with ocular hypertension and glaucoma. They found that H-1337 0.06%, 0.02%, and 0.6% b.i.d. both eyes (O.U.) lowered IOP by 4–5 mmHg on Day 28, 4 h after dosing, whereas the vehicle group lowered IOP by 0.4 mmHg. Treatment-emergent adverse events (TEAEs) occurred in 49% (32/65) of subjects who received H-1337; the most frequent were instillation site erythema, instillation site pain, and conjunctival hyperemia. 13
In the present study, we sought to extend the work of Hartman et al. by evaluating additional concentrations of H-1337 and dosing frequencies (H-1337 ophthalmic solution 0.6% b.i.d., H-1337 ophthalmic solution 1.0% b.i.d., and H-1337 ophthalmic solution 1.0% q.d.) as well as to compare them with a positive control, timolol maleate ophthalmic solution 0.5% b.i.d.
Methods
Study design
This was a Phase 2B, randomized, double-masked, active-controlled, dose–response study conducted at eight private practice sites in the United States. The study was conducted under an Investigational New Drug (IND) in accordance with Good Clinical Practice as required by US Food and Drug Administration regulations. The study was approved by an Institutional Review Board, adhered to the Declaration of Helsinki, and all subjects provided written informed consent before enrollment. This study was registered on ClinicalTrials.gov as NCT05913232.
Masking of q.d. versus b.i.d. was accomplished through the use of “AM” and “PM” labeled bottles. For the q.d. dosing, the PM bottle contained the vehicle of H-1337. Timolol maleate ophthalmic solution 0.5% was acquired from commercial sources. An overwrap and box were used to mask the treatments. Clinical staff and investigators were masked to treatment. An unmasked statistician generated the randomization codes. Other study statisticians, investigators, medical monitor, and other study personnel were masked to the identity of the treatments until all data were entered into the database and locked. H-1337 Ophthalmic Solution, the investigational product (IP), is a benzalkonium chloride-preserved, isotonic, sterile ophthalmic solution buffered at pH 6.5 and was supplied at two concentrations (w/v: 0.6% and 1.0%). Vehicle was identical in formulation to the H-1337 study drug product but without H-1337.
Consenting subjects who met the applicable inclusion/exclusion criteria at the Screening Visit discontinued use of any current ocular hypotensive therapy during the washout period. The washout duration was dependent on the patient’s pre-study ocular hypotensive therapy (based upon previous clinical trial experience with washout periods and recent studies13–15 : muscarinic agonists, oral or topical carbonic anhydrase inhibitors, ≥5 days; β-adrenoceptor antagonists, ≥6 weeks; α-adrenoceptor agonists, ≥5 weeks; prostaglandin analogues, ≥6 weeks; fixed-dose combination products used the component with the longest washout period). The study eye was the eye that met the entry IOP criteria after washout. If both eyes met the criteria, then the study eye was the eye with the higher 8:00 AM IOP at the Baseline Visit. If both eyes had the same 8:00 AM IOP, the study eye was the right eye.
Following the washout period (if applicable), subjects meeting all inclusion/exclusion criteria were randomized to one of four dosing arms in a 1:1:1:1 ratio: H-1337 0.6% b.i.d., 1.0% b.i.d., or 1.0% q.d. (1.0% in the morning with H-1337 vehicle in the evening), and timolol maleate 0.5% b.i.d. The IP was administered b.i.d. O.U. for 28 days.
The study drug was to be taken twice daily (b.i.d.), approximately 12 h apart, at approximately 8 AM and 8 PM, with dosing initiated in the office on Day 1 after the 08:00 h IOP measurement.
Subjects were monitored for safety, tolerability, and efficacy assessments during visits at Baseline (Day 0), Day 1, Day 7, Day 14, and Day 28. At each visit, subjects had safety assessments, measurement of best-corrected visual acuity (BCVA), slit-lamp eye examination, and Goldmann applanation tonometry. Goldmann applanation tonometry was performed to assess IOP at T0 (pre-dose), T0 + 2 h, T0 + 4 h, T0 + 8 h, and T0 + 12 h during the Baseline, Day 1, and Day 28 visits and at T0 on Day 7 and Day 14 visits.
Subject eligibility
Subjects (18 years or older) with a diagnosis of bilateral Primary open-angle glaucoma (POAG) or Ocular hypertension (OHT) were eligible if they had baseline IOP (after washout of any ocular hypotensive drugs) ≥23 mmHg at 8:00 AM in the study eye and ≤32 mmHg in both eyes at all baseline time points. Subjects also were required to meet the following key inclusion criteria: BCVA in both eyes of 20/200 or better Snellen equivalent, and ability to self-administer or have a caretaker administer eye drops. 16 Individuals were excluded if they had closed or very narrow angles or secondary glaucomas, glaucomatous disease severe enough to prevent safe withdrawal from current ocular hypotensive medications, previous glaucoma incisional surgery, recent glaucoma laser or other ocular surgical procedures, clinically significant ocular disease or uncontrolled systemic disease, previous corneal crosslinking or refractive procedures, or prior use of RKI inhibitors resulting in clinically significant adverse events, corneal thickness <480 or >620 µm in the study eye (based upon veracity of Goldmann tonometer), 17 or contraindications to the study medications. Also excluded were subjects with recent ocular corticosteroid use, or concomitant contact lens wear, subjects previously exposed to H-1337, and prior netarsudil use in either eye that resulted in clinically significant adverse events leading to discontinuation of treatment.
IOP assessment
IOP was measured by a calibrated Goldmann applanation tonometer. Two consecutive IOP measurements were taken at each IOP time point, with the right eye being measured first. The applanation probe was withdrawn between measurements. The average of the two measurements was used for analysis. If the two measurements differed by more than 2 mmHg, a third measurement was taken, and the median value was used for analysis. 18
Conjunctival hyperemia assessment
Conjunctival hyperemia was assessed and recorded separately from biomicroscopy through direct visual observation prior to fluorescein instillation and IOP measurement. The hyperemia was graded on a scale of 0–3 using a photographic reference scale and the following guidance: 0 (normal—few vessels of bulbar conjunctiva easily observed); 0.5 (trace)—trace flush, reddish-pink color of the bulbar conjunctiva; 1 (mild)—mild flush, reddish-pink color of the bulbar conjunctiva; 2 (moderate)—bright reddening of the bulbar conjunctiva; 3 (severe)—deep, severe, bright, and diffuse reddening of the bulbar conjunctiva. Subject-reported adverse events were initially evaluated upon subject presentation to the clinic and augmented as needed following the investigator’s observations and examinations.
Statistics
The study was designed to test whether one or more concentrations of H-1337 were non-inferior to the timolol control in the reduction of IOP. As benefit is represented by a negative number, non-inferiority was achieved when the upper limit of the confidence interval (CI) of the difference in treatment (H-1337 minus timolol) was strictly lower than the non-inferiority margin. A sample size of 45 subjects per group allowed for at least 90% power to establish non-inferiority of H-1337 over timolol based on a non-inferiority limit of 2.0 mmHg, a standard deviation (SD) of 3.0 mmHg, and a one-sided t-test (alpha = 0.05). Assuming that 10% of the subjects would not be eligible in the Intent-to-Treat (ITT) Population, a total of 50 subjects per group was required.
A priori, the primary efficacy endpoint selected was change from baseline in IOP (mmHg) at Day 1 and Day 28 (on-treatment time points) in the study eye. The primary population was the ITT group. The endpoints were analyzed using an analysis of covariance model including baseline value and treatment groups as independent variables. The differences between treatments (H-1337 vs. timolol) were calculated for each H-1337 treatment group. A time-matched longitudinal model adjusting for baseline and including a random effect for subject was used to calculate the LS mean, SE, and two-sided 95% CI for the between-treatment differences. Testing for non-inferiority to timolol was conducted in a fixed sequence in hierarchical fashion for 1.0% b.i.d., 0.6% b.i.d., and then 1.0% q.a.m. Additional IOP analyses were also conducted including calculation of mean diurnal IOP (average of each IOP taken during the day), percent decrease in IOP, and proportion of eyes ≤18 mmHg. Nominal P values are reported.
Results
Disposition, demographics, and baseline characteristics
Screened for this study were 248 individuals, of whom 201 subjects were randomized. The demographics of these subjects are presented in Table 1. The mean (SD) age of the overall population was 64.0 (±10.50) years. Overall, 51.7% (104/201) of the population were male, 69.2% (139/201) were White, and 14.4% (29/201) were Hispanic or Latino. All demographics and baseline characteristics appeared largely similar between treatment arms and were typical of controlled studies of ocular hypotensive agents.
Demographics and Baseline Characteristics
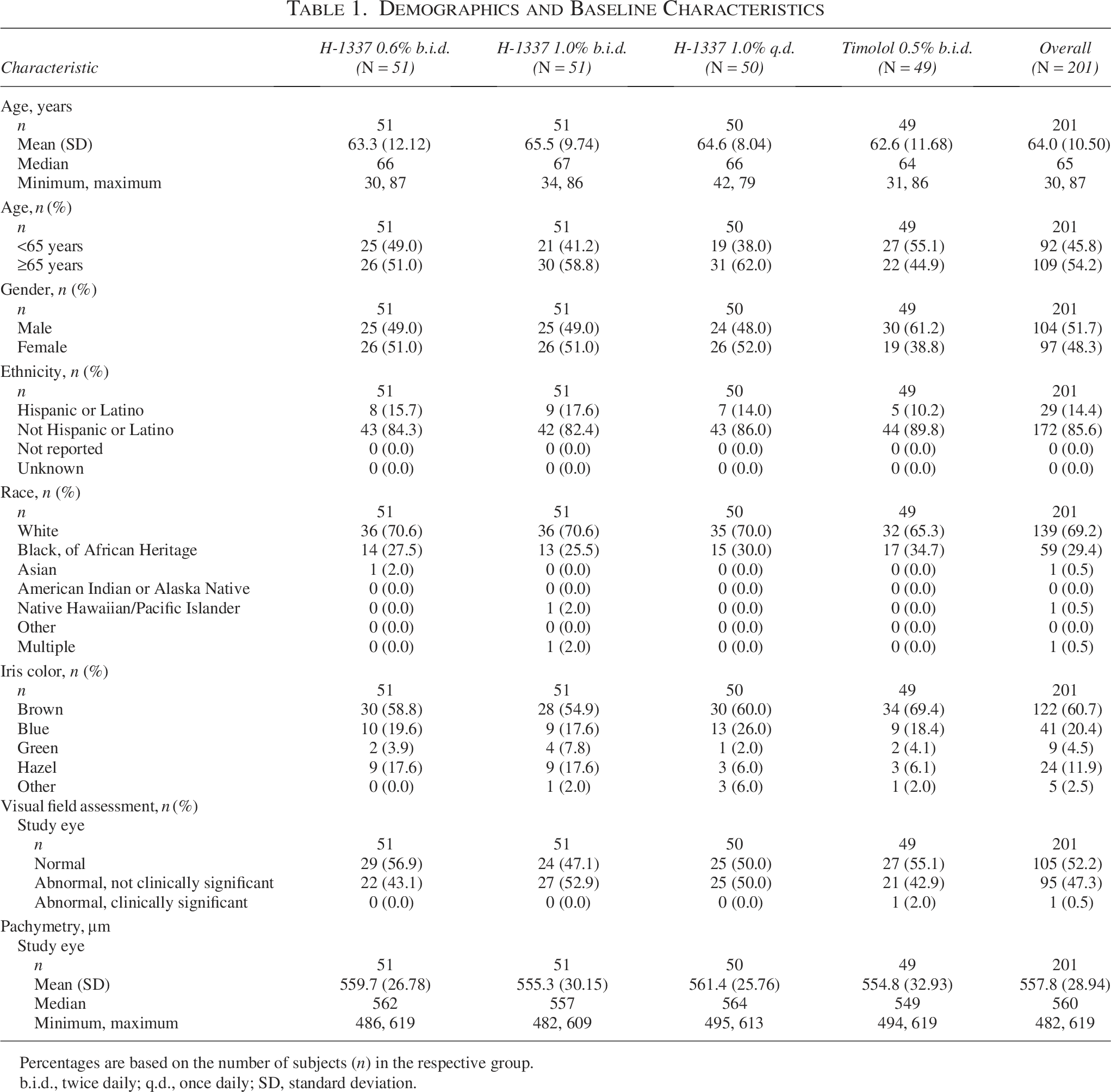
Percentages are based on the number of subjects (n) in the respective group.
b.i.d., twice daily; q.d., once daily; SD, standard deviation.
There were four subjects who discontinued the study due to adverse events: two in the H-1337 1.0% b.i.d. group (instillation site irritation and ocular discharge), one in the H-1337 1.0% q.d. group (epiretinal membrane), and one in the timolol group (bronchospasm).
Of the 201 subjects, at baseline, IOP at +8 h (∼1,600 h) was obtained in all 201 subjects. IOP at +12 h, which was after normal office hours (∼2,000 h), was obtained in 91% (183) of these subjects.
Ocular hypotensive efficacy
On Day 0, after washout of IOP-lowering medications as needed, mean IOPs at 800 h ranged from 25.6 to 26.0 mmHg across treatment arms, with a mean Day 0 12-h diurnal IOP ranging from 23.3 to 23.8 mmHg. Overall, the mean IOPs on Day 0 were similar across the treatment arms. Throughout the study, mean IOP decreased in all treatment groups at all four post-dosing time points on all days tested (P < 0.001; Fig. 1). In general, the ocular hypotensive effects of H-1337 were similar on Day 1 (first dose) and Day 28 (last dose), whereas timolol was more effective on Day 1 than on Day 28.
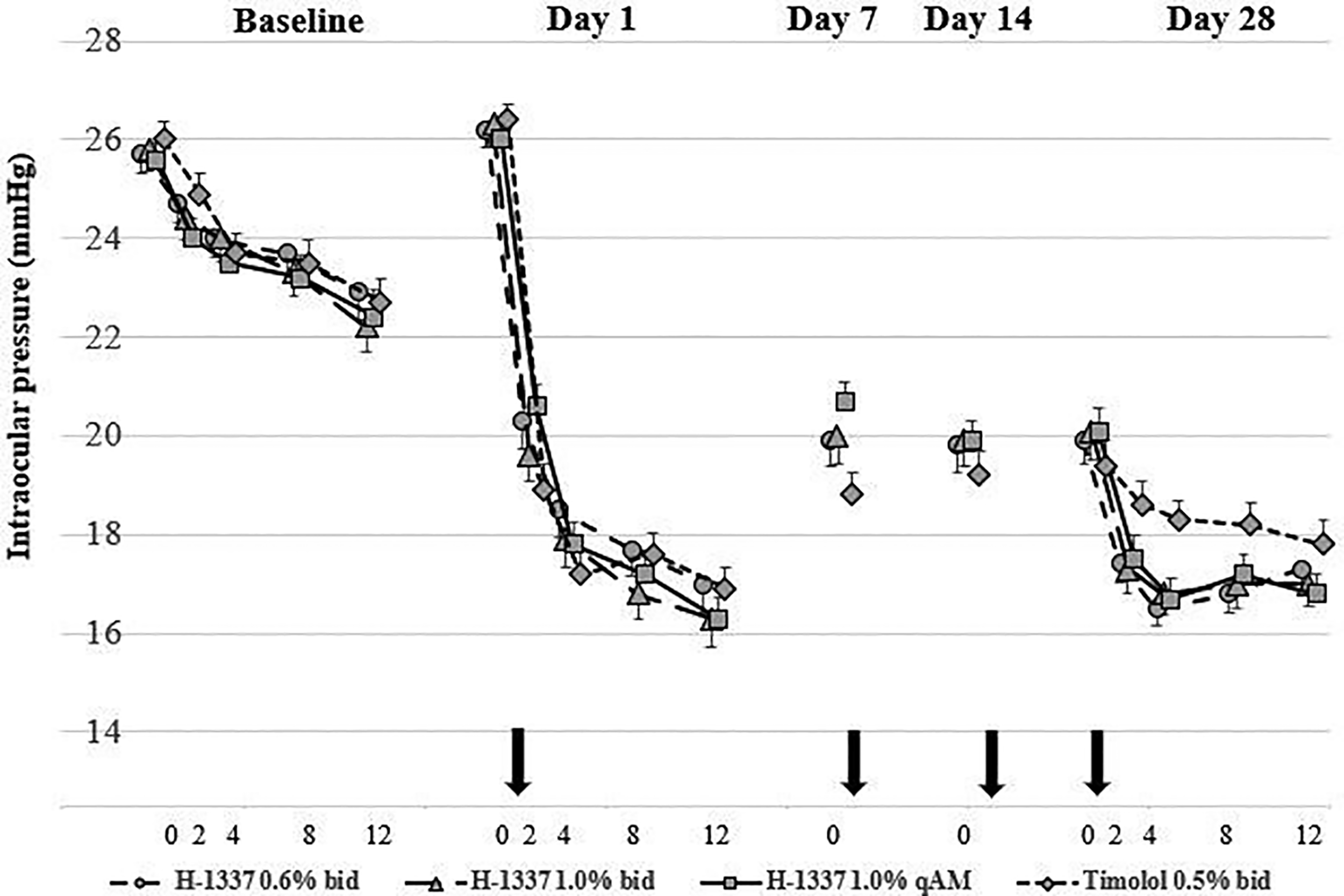
Intraocular pressure: mean (±SEM) (ITT population, mmHg). ITT, intent-to-treat; SEM, scanning electron microscope.
Statistical comparisons among the treatment groups are shown in Table 2. Mean reduction in IOP was 4–7 mmHg for the H-1337 groups and 5–8 mmHg for the timolol group. The difference between H-1337 1.0% b.i.d. and timolol ranged from −1.7 (H-1337 better) to +0.9 mmHg (timolol better) over the 9 time points. Non-inferiority to timolol for H-1337 1.0% b.i.d. (upper limit of 95% CI strictly lower than 1.5 mmHg) was met at 6/9 time points (Day 1—8 and 12 h; Day 28—2, 4, 8, and 12 h). Similar comparative efficacy was seen for the other H-1337 treatment groups.
Intraocular Pressure: Between-Treatment, Time-Matched Comparison of Change from Baseline in Intraocular Pressure in the Study Eye (ITT Population)
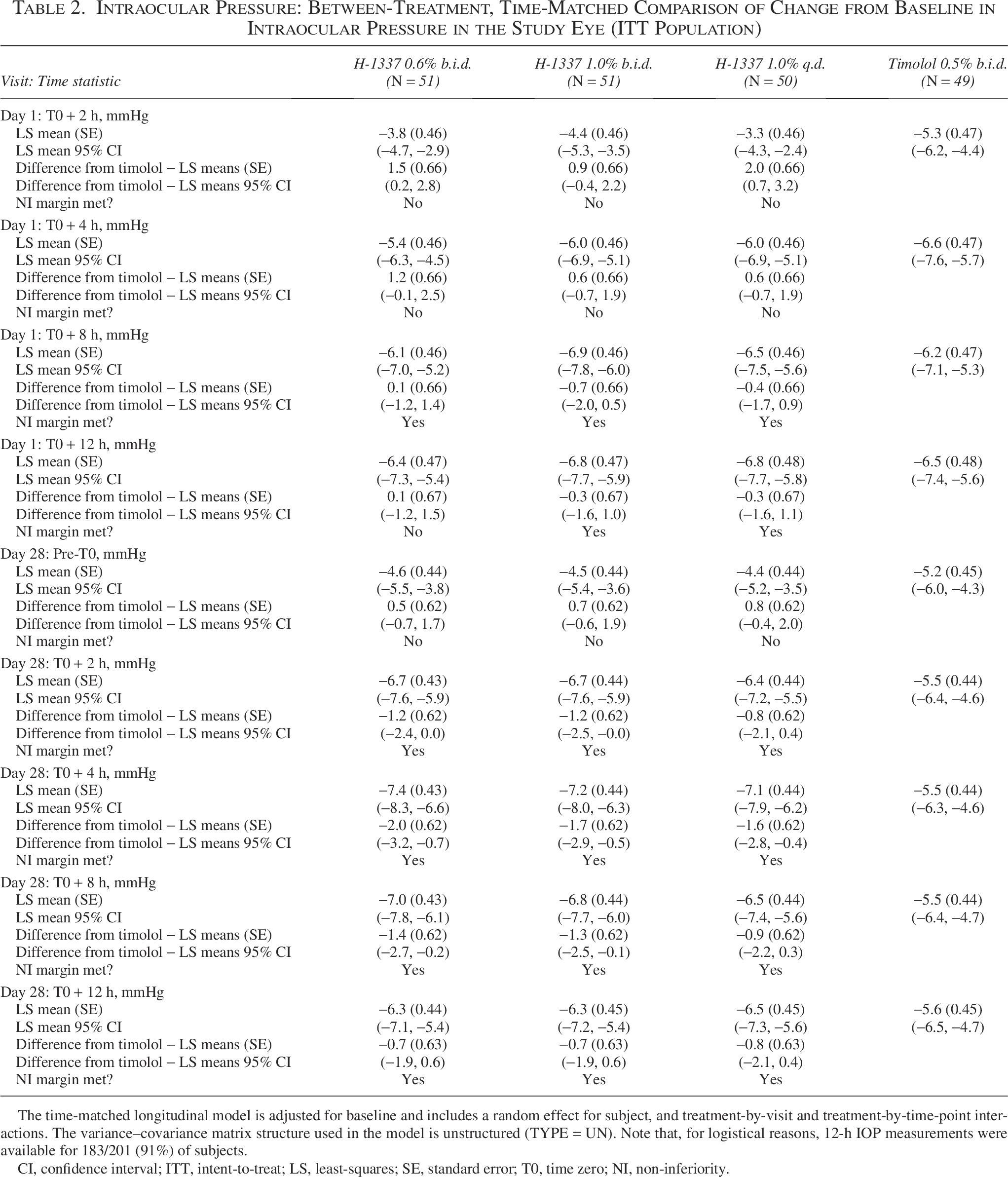
The time-matched longitudinal model is adjusted for baseline and includes a random effect for subject, and treatment-by-visit and treatment-by-time-point interactions. The variance–covariance matrix structure used in the model is unstructured (TYPE = UN). Note that, for logistical reasons, 12-h IOP measurements were available for 183/201 (91%) of subjects.
CI, confidence interval; ITT, intent-to-treat; LS, least-squares; SE, standard error; T0, time zero; NI, non-inferiority.
With respect to the change from baseline in mean diurnal IOP, on Day 1, the least-squares (LS) means difference from timolol for 12-h diurnal IOP was 0.6 mmHg for H-1337 1.0% q.d., 0.7 mmHg for H-1337 0.6% b.i.d., and 0.1 mmHg for H-1337 1.0% b.i.d. On Day 28, the LS means difference from timolol for 12-h diurnal IOP was −1.0 mmHg for H-1337 1.0% q.d., −1.3 mmHg for H-1337 0.6% b.i.d., and −1.2 mmHg for H-1337 1.0% b.i.d. Note that a negative number indicates H-1337 was more effective, and a positive number indicates timolol was more effective.
On Day 1 for the 12-h diurnal period, the proportion of subjects with IOP in the study eye ≤18 mmHg was 53.1% (26/49) in the timolol arm and ranged from 52.0% (26/50) to 54.9% (28/51) in the H-1337 treatment groups. On Day 28, the proportion of study eyes meeting this criterion was 54.2% (26/48) in the timolol arm and ranged from 58.8% (30/51) to 65.3% (32/49) in the H-1337 treatment groups.
The decrease in percent change from the same time on the unmedicated baseline day at each time point in the timolol group ranged from 23% to 27% on Day 1 and 21% to 27% on Day 28. For each of the H-1337 groups, the decrease in percent change was 15% to 28% on Day 1 and 23% to 30% on Day 28.
A post hoc analysis of IOP was conducted on subgroups with baseline (unmedicated) IOP of <25 and ≥25 mmHg; due to the post hoc nature of this analysis, the size in each treatment group in each subgroup was unbalanced—for example, for timolol, <25 mmHg, n = 10, while ≥25 mmHg, n = 39. Nonetheless, H-1337 was shown to be effective in both subgroups but with a larger ocular hypotensive efficacy in subjects with higher baseline IOP levels (reductions of 5.9–8.1 mmHg vs. 3.9–6.5 mmHg on Day 28). Timolol also showed a greater magnitude of effect in subjects with higher baseline IOPs (reductions of 4.9–7.3 mmHg vs. 3.4–5.9 mmHg).
Safety
Adverse events
A total of 115 TEAEs were reported in 66.0% of subjects in the H-1337 1.0% q.d. arm, 78 were reported in 45.1% of subjects in the H-1337 0.6% b.i.d. arm, 107 were reported in 62.7% of subjects in the H-1337 1.0% b.i.d. arm, and 26 were reported in 20.4% of subjects in the timolol arm. Of the 326 TEAEs reported in this study, 11 were non-ocular (4 were related to IP) and 315 were ocular in nature (305 were related to IP). The most commonly reported TEAEs among subjects in each treatment arm were conjunctival hyperemia (H-1337 arms, 40.1%; timolol arm, 8.2%) and instillation site irritation (H-1337 arms, 16.4%; timolol arm, 6.1%; Table 3). No TEAEs were classified as serious or led to death. As noted above, four subjects had TEAEs that led to IP discontinuation and study withdrawal. The overall rate of hyperemia (conjunctival and/or ocular) among subjects was 54.0% (27/50) for H-1337 1.0% q.d., 33.3% (17/51) for H-1337 0.6% b.i.d., 41.2% (21/51) for H-1337 1.0% b.i.d., and 8.2% (4/49) for timolol.
Summary of Treatment-Emergent Adverse Events by System Organ Class and Preferred Term
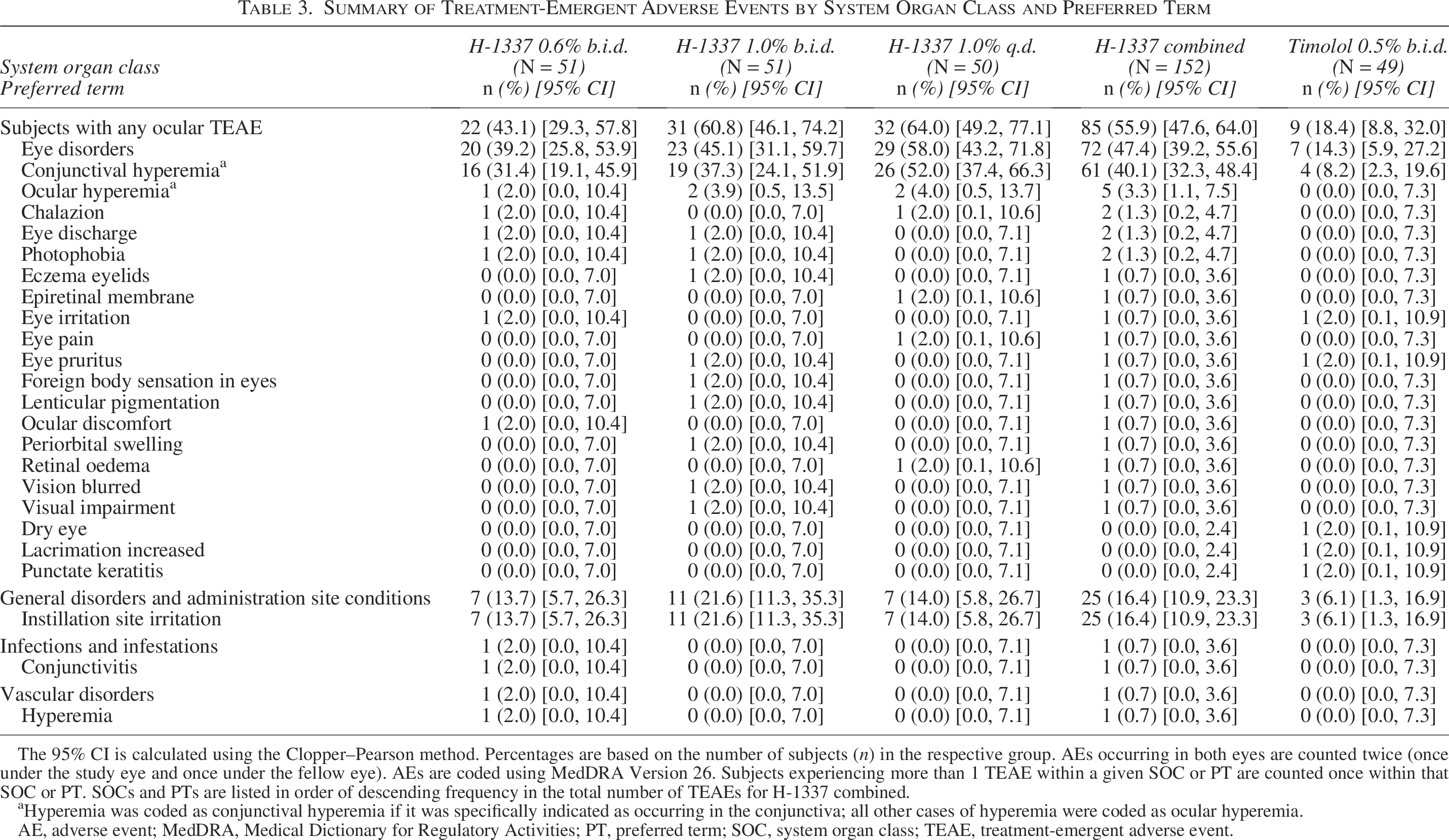
The 95% CI is calculated using the Clopper–Pearson method. Percentages are based on the number of subjects (n) in the respective group. AEs occurring in both eyes are counted twice (once under the study eye and once under the fellow eye). AEs are coded using MedDRA Version 26. Subjects experiencing more than 1 TEAE within a given SOC or PT are counted once within that SOC or PT. SOCs and PTs are listed in order of descending frequency in the total number of TEAEs for H-1337 combined.
aHyperemia was coded as conjunctival hyperemia if it was specifically indicated as occurring in the conjunctiva; all other cases of hyperemia were coded as ocular hyperemia.
AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities; PT, preferred term; SOC, system organ class; TEAE, treatment-emergent adverse event.
All conjunctival hyperemia TEAEs were related to the study drug, and the incidence of these TEAEs was not dose-related in the H-1337 arms (1.0% q.d., 52.0%; 0.6% b.i.d., 31.4%; 1.0% b.i.d., 37.3%). The majority of subjects with conjunctival hyperemia TEAEs in the H-1337 0.6% and 1.0% b.i.d. arms had a maximum of mild severity, whereas the subjects with conjunctival hyperemia TEAEs in the H-1337 1.0% q.d. and timolol arms were evenly split between mild and moderate/severe intensity.
Shown in Fig. 2 is the distribution of severity grades of conjunctival hyperemia as judged by the investigators at the slit lamp for four time points—Day 1 +4 h, Day 1 +12 h, Day 28 +4 h, and Day 28 +12 h. Hyperemia was seen in subjects in all treatment groups. In subjects with hyperemia, the severity was trace, mild, or moderate for nearly all subjects. The severity was moderate in approximately one-third of the subjects in the H-1337 groups at 4 h after dosing on Day 1, reducing in severity throughout the first day of dosing and with longer treatment duration.
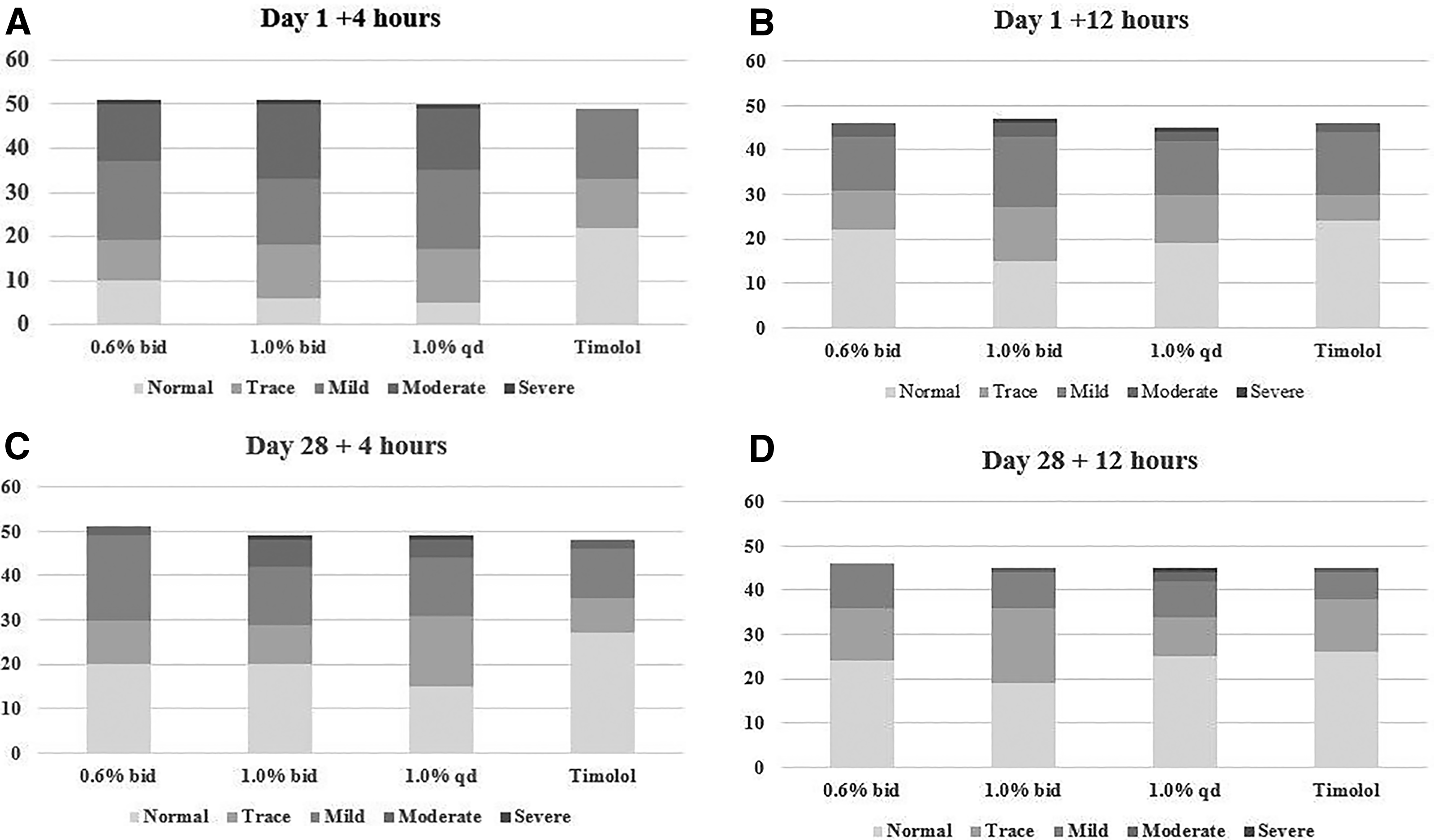
Severity of conjunctival hyperemia: number of subjects per category.
Changes in blood pressure were minor and consistent across all arms of the study. Heart rate tended to decrease in all arms of the study. On Day 28, decreases in heart rate were greater in the timolol arm than in any of the H-1337 arms.
There were no clinically significant changes in visual acuity or ophthalmoscopy as judged by the investigators.
Discussion
The primary objective of this Phase 2B study was to evaluate the ocular hypotensive efficacy of three dose regimens of H-1337 compared with timolol maleate 0.5% in subjects with POAG or OHT treated for up to 28 days. We purposefully selected a high hurdle for this study—the US regulatory requirement for Phase 3 of non-inferiority to timolol at 1.5 mmHg at multiple time points—in order to assess the potential for later-stage development, even though the sample size for this study was much smaller than typical for a Phase 3 study (∼50 rather than ∼150). For H-1337 1.0% b.i.d. (the highest dose), this non-inferiority margin was met at 5 time points—Day 1 +8 h, and on Day 28 at +2, +4, +8, and +12 h. Although further hierarchical comparisons were technically unnecessary, an examination of the other two active arms (H-1337 1.0% q.d. and 0.6% b.i.d.) showed six of the nine comparisons met the 1.5 mmHg threshold for non-inferiority.
The mean differences between H-1337 and timolol ranged from −2.0 (H-1337 better) to +1.5 (timolol better). This information will be useful in planning required sample sizes for potential future studies.
Consistent with the pharmacology of this molecule, the most common treatment-related TEAEs among subjects in each treatment arm were conjunctival or ocular hyperemia and instillation site irritation. These were typically of mild severity and short duration.
Of note, there was little dose–response in either ocular hypotensive efficacy or hyperemia among the three doses of H-1337. For pharmaceutical and nonclinical safety reasons, 1.0% is most likely the maximal concentration of H-1337 that may be used.
The results from the present study are consistent with the previous study conducted by Hartman et al. In that study, the dose–response curve was relatively flat, although 0.6% b.i.d. was slightly more effective than 0.06% and 0.2% H-1337 b.i.d. There was little dose–response in ocular hyperemia in that study as well.
The ocular hypotensive efficacy of timolol in the present study was less on Day 28 than on Day 1. While one might conclude a tolerance developing to timolol, we suggest that this might just be random variability, as it is inconsistent with another recent study using timolol as a positive control. 6 The incidence of hyperemia was also slightly greater in the 1.0% H-1337 q.d. group than in the 1.0% H-1337 b.i.d. group. Again, we exercise caution in overinterpreting this and suggest it was due to variability. Future larger studies might be needed to evaluate this observation.
In conclusion, in this Phase 2, double-masked, active-controlled study, H-1337 in doses of 0.6% b.i.d., 1.0% q.d., and 1.0% b.i.d. had ocular hypotensive efficacy in the range of timolol, although not within the strict Phase 3 non-inferiority margins, which would have required a larger sample size. The primary AE, conjunctival hyperemia, is consistent with the pharmacology of this molecule and was observed in approximately 40% of subjects treated with H-1337. However, the hyperemia was mild in intensity and led to only one discontinuation of treatment. Selection of optimal dosing may require longer-term evaluation in larger trials.
Authors’ Contributions
L.S.: Methodology, investigation, writing (original draft, review and editing). J.M.: Methodology, investigation, writing (original draft, review and editing). R.D.W.: Methodology, conceptualization, formal analysis, writing (original draft, review and editing). J.B.: Methodology, formal analysis, data curation, writing (original draft, review and editing), supervision. J.A.S.: Methodology, formal analysis, data curation, writing (original draft, review and editing), supervision, project administration. G.D.N.: Methodology, conceptualization, formal analysis, writing (original draft, review and editing), supervision, project administration.
Footnotes
Acknowledgments
The authors thank Jennifer Klem, PhD of Klem Medical Communications, LLC for medical writing, and Esmeralda Meunier, MSc for biostatistical support. The H-1337 Phase 2B investigators were: Douglas G. Day, MD, Roswell, GA; El-Roy D. Dixon, MD, Albany, GA; James H. Burden, Jr, MD, Colorado Springs, CO; Sherif M. El-Harazi, MD, MPH, Glendale, CA; Paul J. Hartman, MD, Rochester, NY; Jay Mulaney, MD, Lakeland, FL; Kenneth W. Olander, MD, PhD, Maryville, TN; Philip L. Shettle, DO, Largo, FL.
Author Disclosure Statement
L.S.: Clinical Study Grant, DWTI. J.M.: Clinical Study Grant, DWTI. R.D.W.: Consultant to several medical device and pharmaceutical firms. J.B.: None. J.A.S.: Consultant to several medical device and pharmaceutical firms. G.D.N.: Consultant to several medical device and pharmaceutical firms.
Funding Information
This study was supported by D. Western Pharmaceuticals Institute, Inc.