
Abstract
Purpose:
Cetalkonium chloride (CKC) is a cationic agent used in ophthalmical emulsions. Despite its growing application in ocular drug delivery, its safety profile on corneal endothelial cells remains unclear. This study evaluated the in vitro cytotoxic effects of CKC on human corneal endothelial cells (HCEnCs).
Methods:
HCEnCs were exposed to CKC at concentrations ranging from 0.03125 to 4.0 × 10−4% (w/v) for 24–72 h. Cell viability was assessed using Cell Counting Kit-8 and lactate dehydrogenase (LDH) assays. Live/dead cell staining was performed for morphological confirmation. Reactive oxygen species (ROS) production and mitochondrial function were evaluated using DCFDA and MitoTracker assays. Western blot analysis was conducted to examine CKC-induced changes in cell survival pathways, including mammalian target of rapamycin (mTOR), protein kinase B (Akt), extracellular signal-regulated kinase (ERK), Bcl-2-associated X protein (BAX), and B-cell lymphoma-extra-large (Bcl-xL).
Results:
CKC induced dose- and time-dependent cytotoxicity in HCEnCs. Exposure to CKC at concentrations ≥0.25 × 10−4 % for over 48 h significantly reduced cell viability and increased LDH release and ROS production. At concentrations ≥1.0 × 10−4 %, cell viability was reduced by more than 50% at both 48 and 72 h. In surviving cells, mitochondria showed minimal structural alterations. CKC exposure inhibited cell survival pathways such as mTOR, Akt, Bcl-xL, and ERK, while the proapoptotic pathway marker BAX was upregulated.
Conclusion:
CKC exhibits dose- and time-dependent toxicity in HCEnCs, mediated by oxidative stress and the modulation of survival and apoptotic signaling pathways. However, it is challenging to directly extrapolate laboratory conditions to the clinical setting. Therefore, these findings should be interpreted with caution, particularly in scenarios where direct exposure of the corneal endothelium to CKC-containing formulations is anticipated.
Introduction
The cornea is the most important tissue forming the surface of the eye, consisting of the corneal epithelium, keratocytes, and corneal endothelial cells. 1 The corneal epithelium has a remarkable regenerative capacity, with all epithelial cells repopulating within 1–2 weeks. 2 In contrast, the corneal endothelium, a single layer of cells located at the innermost part of the cornea, is known to lack regenerative capability after birth. 3 Damage to the endothelial cell layer results in irreversible corneal edema and opacity, leading to a condition known as bullous keratopathy. 4
Ophthalmical medications often contain preservatives to prevent bacterial contamination. 5 Among these, quaternary ammonium compounds like benzalkonium chloride (BAK) are widely used in eye drops due to their broad-spectrum antimicrobial properties, effectively targeting various microorganisms, including gram-positive and gram-negative bacteria and fungi.5,6 BAK remains the most commonly used preservative in ophthalmology; however, concerns over its potential ocular toxicity have driven research into alternative compounds.5,7–9 One such substitute is cetalkonium chloride (CKC), another quaternary ammonium compound with a C16 alkyl chain length. 10
CKC is sometimes present in minimal amounts within BAK mixtures and has been found to accumulate in the lipid layer of the tear film model, where it enhances stability in a concentration-dependent manner. 10 In contrast, BAK interacts with the lipid layer in a way that compromises the tear film’s integrity. CKC has been incorporated into cationic-emulsion eye drops, such as those containing latanoprost or cyclosporine nano-emulsion. 11 Its positive charge improves the bioadhesive properties of these emulsions, facilitating better adherence to the negatively charged ocular surface and enhancing the penetration of active ingredients into ocular tissues. 10
Previously, we reported that CKC exposure induces significant toxicity in human corneal epithelial cells. 12 Our findings show that at concentrations starting from 1.0 × 10−4 %, over 50% of corneal epithelial cells died within 48 h. At 2.0 × 10−4 %, CKC exposure led to 90% cell death after 24 h and nearly complete cellular death after 48 h. These results suggest the need to limit CKC concentration in eye drops to levels below ocular toxicity.
Ophthalmical drugs applied to the ocular surface inevitably interact with corneal cells as they permeate the cornea to reach intraocular target tissues, making contact with corneal endothelial cells unavoidable. Given the limited regenerative capacity of these cells, it is crucial to evaluate the safety of CKC, a compound used in approved ophthalmical emulsions, whose direct impact on the nonregenerative corneal endothelium remains unknown.
This study was designed to investigate the potential cytotoxic effects of CKC on cultured human corneal endothelial cells (HCEnCs). Given the structural similarity between CKC and BAK, we aimed to explore whether CKC exerts comparable toxic effects on HCEnCs. Understanding the impact of CKC on cell viability, oxidative stress, and mitochondrial integrity is essential for evaluating its safety in ophthalmical formulations, particularly considering the nonregenerative nature of corneal endothelial cells.
Methods
HCEnCs culture
The HCEnC line, B4G12 (Cat no. CSC-C3457-CRA, Shirley, NY, USA) was obtained from Creative Bioarray (Shirley, NY, USA). Cells were maintained in the recommended human endothelial serum-free medium (Creative Bioarray, Cat no. CM-345L7, Shirley, NY, USA) supplemented with 10 ng/mL fibroblast growth factor-2 (Creative Bioarray, Cat no. CSC-CTK0134, Shirley, NY, USA). The culture medium was replaced every 3 days, and cells were subcultured using 0.25% Trypsin-EDTA (Gibco BRL, Carlsbad, CA, USA).
Preparation of CKC
CKC), obtained from Merck KGaA (Darmstadt, Germany), was dissolved in methanol (Merck) for serial dilution, resulting in a vehicle concentration of 0.004% (v/v) methanol. 12 A 10% (w/v) stock solution was prepared, and fresh CKC working solutions were utilized for each experiment.
Cell viability assay
The viability of HCEnCs was evaluated using the Cell Counting Kit-8 (CCK-8) from Dojindo Laboratories (Kumamoto, Japan), as described in a previous study. 12 Cells were plated in a 96-well plate at a density of 1 × 104 cells per well and allowed to adhere. Once confluency reached 80%, CKC was then administered at concentrations of 0, 0.03125, 0.0625, 0.125, 0.25, 0.5, 1.0, 2.0, and 4.0 × 10−4 % (w/v) for incubation periods ranging from 24 to 72 h. 12 Following treatment, 10 μL of CCK-8 reagent was introduced into each well, and the plates were incubated at 37°C for an additional 4 h. Absorbance at 450 nm was then measured using a microplate reader to determine cell viability.
Live and dead cell staining
To assess both quantitative and qualitative aspects of cell viability and cytotoxicity in CKC-treated HCEnCs, a live/dead viability/cytotoxicity kit (Molecular Probes, Cat. L3224, Thermo Fisher Scientific, Rochester, NY, USA) was utilized as described in a previous study. 12 HCEnCs were cultured in confocal dishes and exposed to CKC at concentrations of 0.25, 0.5, 1.0, and 2.0 × 10−4 % (w/v) for 24 h. Prior to staining, cells were washed twice with DPBS, and staining reagents—calcein AM (final concentration: 2 μM) and EthD-1 (final concentration: 4 μM)—were prepared in DPBS according to the manufacturer’s guidelines. The stained cells were then incubated at 37°C for 30 min in a dark environment to minimize photobleaching. Following incubation, excess stain was removed via gentle DPBS washes, and fluorescence imaging was performed using a confocal live imaging system (Leica Microsystems CMS GmbH, Mannheim, Germany).
Measurement of reactive oxygen species
Intracellular reactive oxygen species (ROS) levels were assessed using the dichlorofluorescin diacetate (DCFDA)/H2DCFDA-Cellular ROS Assay Kit (Abcam, Cat. ab113851; Cambridge, UK) as described in a previous study. 12 HCEnCs were treated with CKC at concentrations of 0, 0.0156, 0.0313, 0.0625, 0.125, 0.25, 0.5, 1.0, and 2.0 × 10−4 % (w/v) for 20 min. Following exposure, the culture medium was discarded, and cells were washed with 100 µL/well of 1× ROS buffer, following the manufacturer’s instructions. Staining was performed in the dark environment at 37°C by adding 100 µL/well of a 20 µM solution of 2′,7′-DCFDA, followed by a 45-min incubation period. Subsequently, cells were gently washed with 100 µL/well of 1× ROS buffer to remove excess stain. Fluorescence measurements were immediately recorded at 485 nm excitation/535 nm emission using an endpoint assay.
Lactate dehydrogenase assay
Cellular toxicity under different experimental conditions was assessed using a colorimetric assay with the lactate dehydrogenase (LDH) cytotoxicity detection kit (Takara Bio Inc., Shiga, Japan) as described in a previous study, following the manufacturer’s instructions. 12 The LDH assay quantitatively measures LDH released from nonviable cells. HCEnCs were seeded in a 96-well plate at a density of 1 × 104 cells per well and allowed to adhere. Once confluence reached 80%, cells were then exposed to CKC at concentrations of 0.03125, 0.0625, 0.125, 0.25, 0.5, 1.0, 2.0, and 4.0 × 10−4 % (w/v) for incubation periods of 24, 48, and 72 h. After each incubation, LDH activity was measured in the collected supernatants, with 10% (v/v) LDH solution added. Wells without CKC served as negative controls, while wells treated with 1% Triton X-100 functioned as positive controls. Absorbance was recorded at 490 nm to determine cytotoxicity levels.
Mitotracker assay
To assess mitochondrial activity and localization in live cells, MitoTracker Deep Red FM (Molecular Probes, Cat. M22426) was used as a mitochondria-selective probe suitable for subsequent immunostaining as described in a previous study. 12 HCEnCs were seeded in 4-well Nunc Lab-Tek II chamber slides (Thermo Fisher Scientific) at a density of 4 × 104 cells/mL and treated with CKC for 24 h. Following treatment, the cells were incubated with MitoTracker solution (final concentration: 100 nM) at 37°C for 30 min in the dark to minimize photobleaching. After incubation, excess dye was removed by gentle washing with DPBS, and cells were fixed with 10% formaldehyde at room temperature (RT) for 10 min. 12 Permeabilization was performed using 0.1% Triton X-100 for 5 min at RT, followed by DPBS rinsing. To block nonspecific binding, 1% bovine serum albumin (BSA) in DPBS was applied for 30 min at RT. Slides were then incubated overnight at 4°C with Goat anti-Zo1 (1:200; Abcam, Cat. ab190085), washed with DPBS, and further incubated with Alexa488-conjugated donkey anti-goat antibody (1:1000; Abcam, Cat. ab150129) at RT for 1 h. 12 After multiple washes, Tetramethylrhodamine isothiocyanate-conjugated phalloidin (1 µg/mL; Sigma-Aldrich) was used to stain F-actin, and nuclei were counterstained with 4ʹ,6-diamidino-2ʹ phenylindole (DAPI; Roche, Cat. 10236276001; Mannheim, Germany).
Western blot assay for cell survival signals
CKC-treated cells (for 24 h) were lysed in ice-cold radioimmunoprecipitation assay buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, and 0.1% sodium dodecyl sulfate [SDS]) for 30 min. 12 Cellular debris was removed by centrifugation at 16,000g for 10 min. A total of 20 μg of protein from each sample was separated using SDS polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Millipore Corporation, Billerica, MA). Membranes were blocked for 1 h at room temperature in TTBS buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween 20) containing 3% BSA. Following blocking, membranes were incubated overnight at 4°C with primary antibodies, including rabbit anti-mTOR (1:1,000; Cell Signaling; Cat. 5536; Danvers, MA, USA), rabbit anti-phospho-mTOR (1:1,000; Cat. 2983; Cell Signaling), rabbit anti-Akt (1:1,000; Cell Signaling; Cat. 9272), rabbit anti-phospho-Akt (1:1,000; Cell Signaling; Cat. 4060), rabbit anti-P-p44/42 MAPK (extracellular signal-regulated kinase [ERK] 1/2) (1:1,000; Cell Signaling; Cat. 4370), rabbit anti-p44/42 MAPK (ERK 1/2) (1:1,000; Cell Signaling; Cat. 4695), rabbit anti-BAX (1:1,000; Cell Signaling; Cat. 2772), rabbit anti-B-cell lymphoma-extra-large (Bcl-xL) (1:1,000; Cell Signaling; Cat. 2764), and mouse anti-β-actin (1:50,000; Sigma-Aldrich; Cat. A5441). 12 Secondary antibody incubation with horseradish peroxidase-conjugated antibodies was carried out at room temperature for 1 h. Signal detection was performed using Pierce enhanced chemiluminescence substrate (Thermo Fisher Scientific; Cat. 32106) and visualized with a Fusion Pulse 6 chemiluminescence system (Vilber Lourmat, Marne-la-Vallee, France). Densitometric analysis was conducted using Multi Gauge V3.0 software (Fujifilm Life Science, Tokyo, Japan). All experiments were performed at least in triplicate.
Statistical analysis
The data are expressed as the mean ± standard error. Statistical significance was assessed using analysis of variance, followed by Dunnett’s multiple comparison test as described in the previous study. 12 A P value less than 0.05 was considered statistically significant. All analyses were conducted using GraphPad Prism Ver. 9.3.1 (GraphPad Software Inc., La Jolla, CA, USA).
Results
Cell viability after exposure to CKC
Cell viability analysis demonstrated a dose- and time-dependent decline in HCEnCs viability after CKC exposure. At concentrations ≤0.125 × 10−4 %, no significant decrease in viability was observed at 24 h; however, prolonged exposure to the same concentration at 48 and 72 h resulted in notable toxicity. CKC concentrations ≥0.25 × 10−4 % reduced viability significantly at 24 h. At 1.0 × 10−4 %, cell viability was reduced by more than 50% at 48 and 72 h, while exposure to concentrations ≥2.0 × 10−4 % resulted in over 75% viability loss at 24 h and exceeded 90% cell death at 48 and 72 h (Fig. 1A). To further validate CKC-induced cytotoxicity, LDH assays were conducted to assess membrane integrity. Exposure to high CKC concentrations led to a substantial increase of LDH, confirming the dose-dependent toxicity previously observed in the CCK8 assay. The increased LDH release correlates with the rapid loss of cell viability, particularly at CKC concentrations exceeding 1.0 × 10−4 % (Fig. 1B). Live/dead cell staining performed after 24 h of exposure revealed a clear dose-dependent shift in the proportions of viable and dead cells (Fig. 2). At lower CKC concentrations, most cells retained viability; however, increasing CKC exposure resulted in a progressive reduction in live cells, further substantiating the toxic effects observed in the CCK8 and LDH assays.
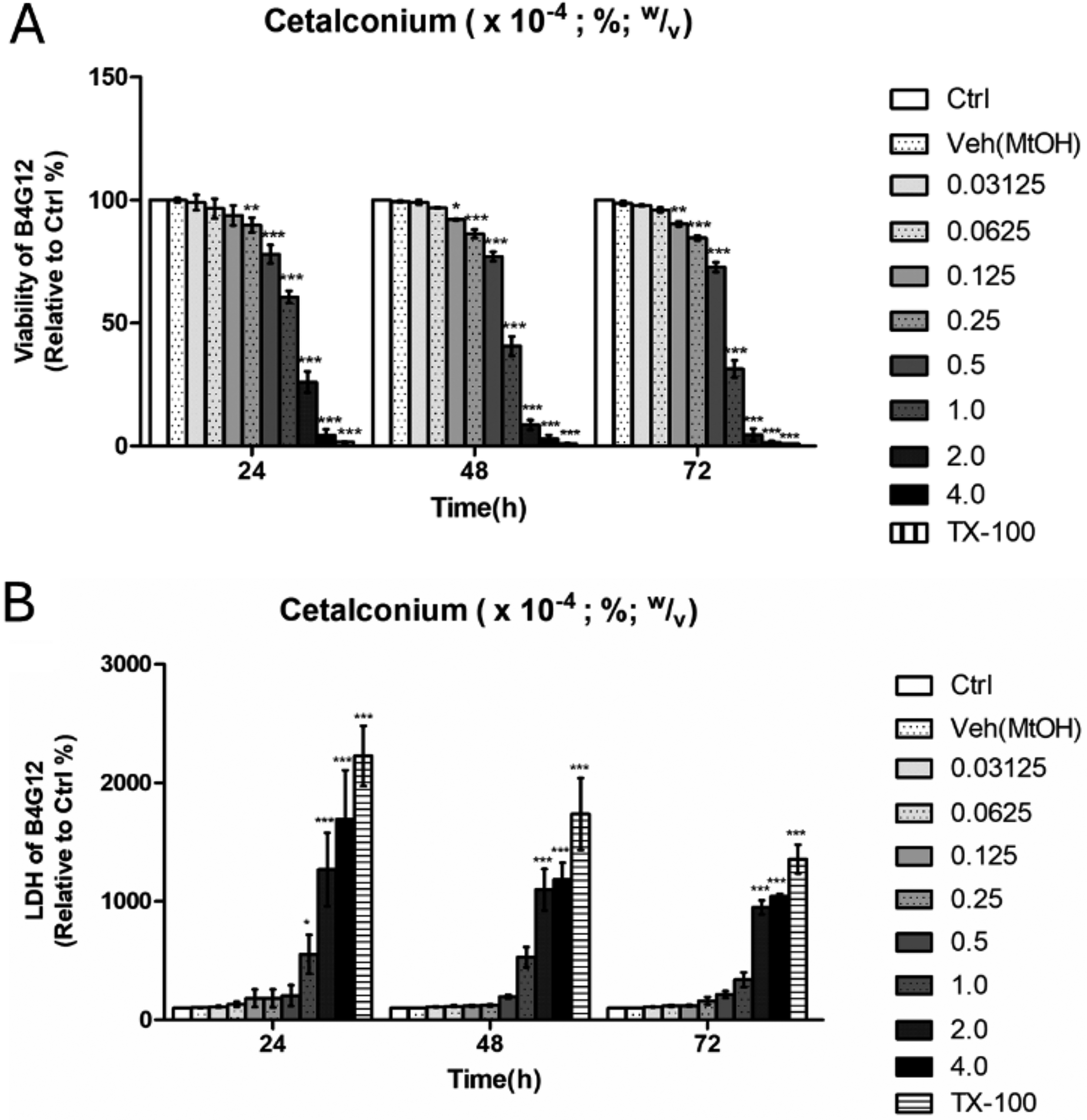
Viability change of HCEnCs after exposure to CKC.
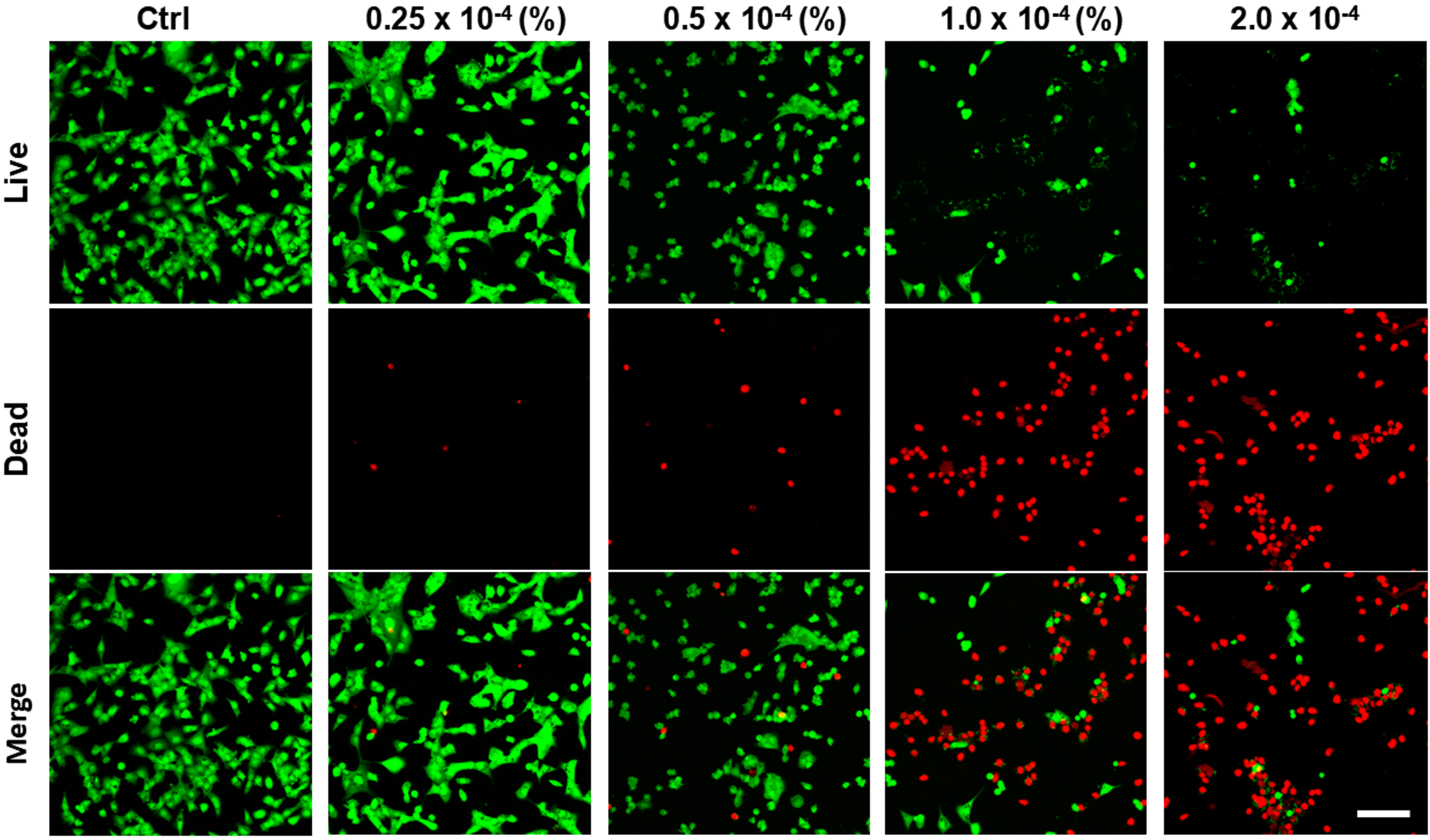
Live/dead cell analysis of HCEnCs after exposure to CKC. Live/dead cell staining after 24 h of exposure showed a dose-dependent shift in live and dead cell populations. Scale bar: 100 µm.
Mitotracker assay and ROS assay
Mitochondrial structure and localization showed no significant morphological changes in surviving cells compared to control cells, suggesting that mitochondrial integrity was preserved at the tested concentrations. However, exposure to higher CKC concentrations led to a pronounced reduction in the overall cell population, indicating substantial cytotoxicity. Importantly, the assessment of mitochondrial morphology was limited to surviving cells, which retained mitochondrial features comparable to those of untreated controls. These findings imply that CKC-mediated toxicity primarily affects cell viability rather than directly disrupting mitochondrial architecture (Fig. 3A). To assess oxidative stress induced by CKC, an acute ROS assay was performed by measuring ROS levels in HCEnCs following a 20-min exposure to CKC. The results revealed a concentration-dependent increase in ROS generation, indicating that CKC exposure disturbs redox homeostasis within the cells (Fig. 3B). These findings suggest that CKC induces oxidative stress in HCEnCs, which may contribute to its cytotoxic effects. To further evaluate CKC-induced cellular changes, mitochondrial morphology and distribution were analyzed in HCEnCs exposed to CKC.
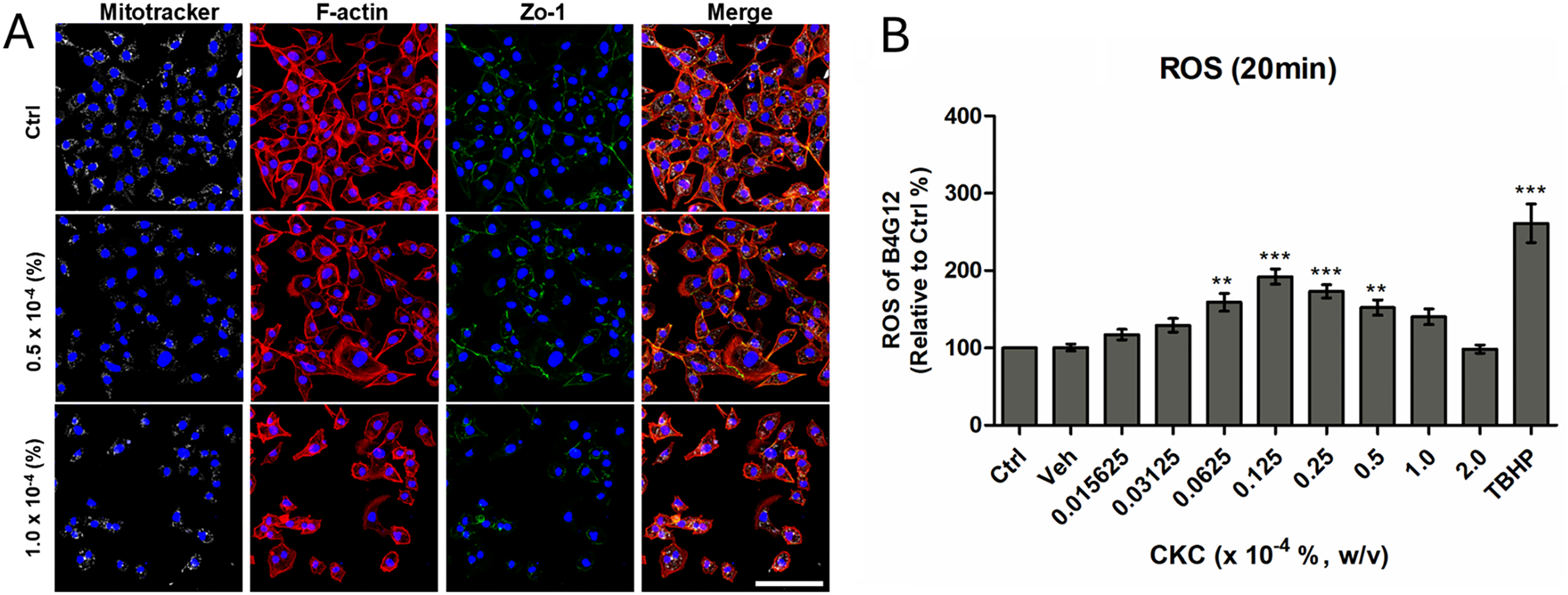
Reactive oxygen species and Mitotracker assay of HCEnCs after exposure to CKC.
Effect of CKC on HCEnCs survival pathway
Exposure to CKC resulted in the suppression of mTOR, ERK, and Akt phosphorylation, which are key regulators of cell division and migration in HCECs. This inhibitory effect became more pronounced as CKC concentration increased. Additionally, CKC exposure led to a reduction in Bcl-xL expression, a protein that inhibits apoptosis, while concurrently upregulating BAX, a pro-apoptotic factor. Consequently, the BAX/Bcl-xL ratio increased in a dose-dependent manner following CKC treatment (Fig. 4).
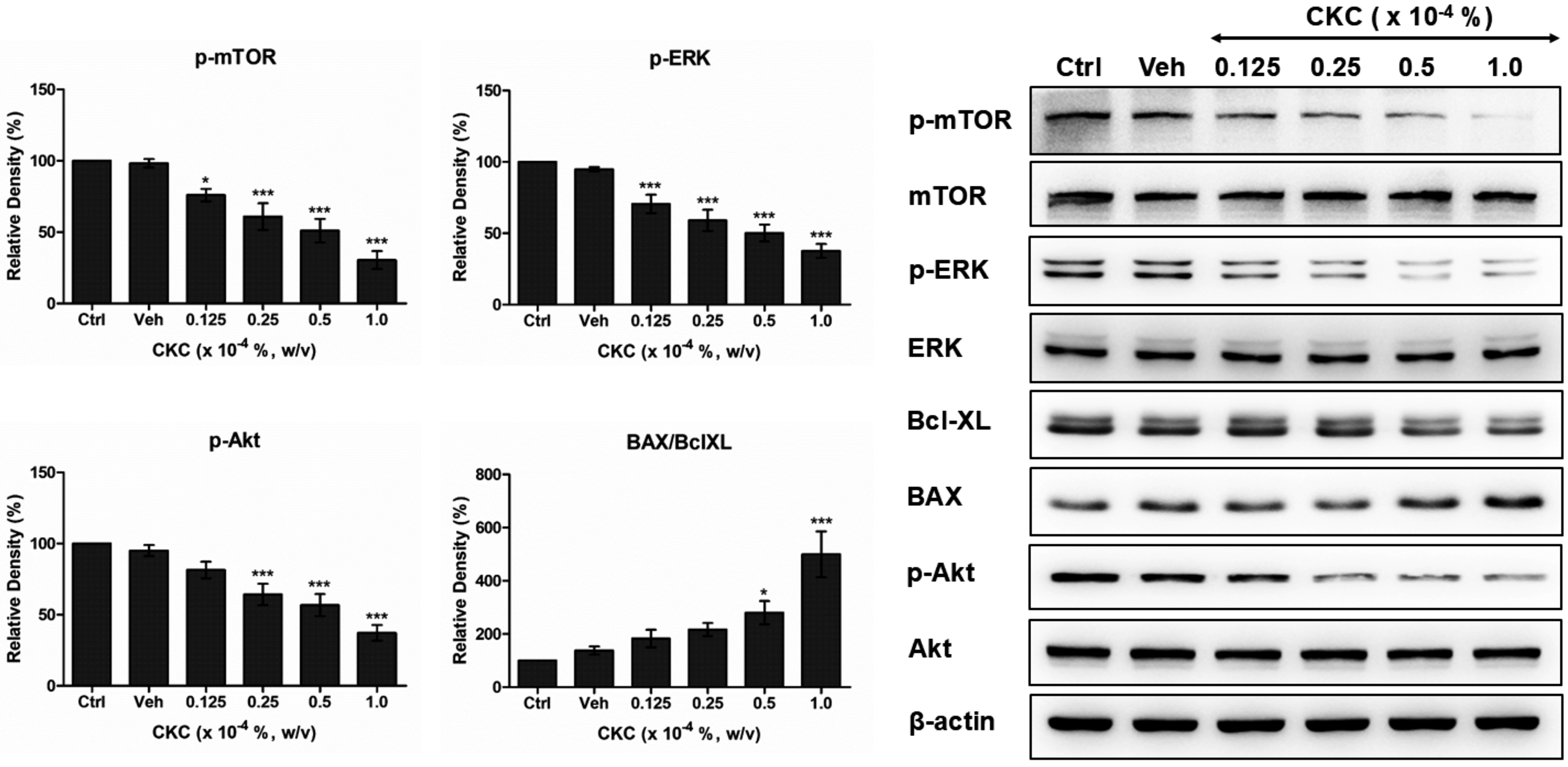
Effect of CKC on the cell survival pathway of HCEnCs. The expression levels of cellular survival signals such as phosphorylated mTOR, phosphorylated Akt, phosphorylated ERK, and Bcl-xL demonstrated a dose-dependent decrease in HCEnCs after exposure to CKC for 24 h, accompanied by increased expression pro-apoptotic signal of BAX. Ctrl: cells cultured in media only (negative control). Veh: culture media containing 0.004% (v/v) methanol (vehicle control). Values are presented as mean ± SEM and were obtained from at least three independent experiments; One-way ANOVA showed a significant treatment effect (p-mTOR: F(5,30) = 20.09; pAKT: F(5,30) = 17.80; p-ERK: F(5,42) = 23.29; BAX/BclXL: F(5,36) = 10.97, P < 0.0001). Dunnett’s post hoc test was used to compare each treatment group to the control. *P < 0.05, **P < 0.01, ***P < 0.001. Akt, protein kinase B; BAX, Bcl-2-associated X protein; Bcl-XL, B-cell lymphoma-extra-large; ERK, extracellular signal-regulated kinase; mTOR, mammalian target of rapamycin.
Discussion
In this study, we observed that CKC induced significant toxicity in HCEnCs in a dose- and time-dependent manner. Specifically, exposure to CKC concentrations of 1.0 × 10−4% or higher resulted in more than 50% toxicity, while concentrations of 2.0 × 10−4% led to over 90% of cell death after 48 h. This finding aligns with previously reported CKC toxicity in corneal epithelial cells. 12
CKC has recently been used in ophthalmical formulations to create cationic emulsions, which offer several advantages for ocular drug delivery.11,13 These emulsions enhance retention by leveraging their positive charge, which interacts with the negatively charged ocular surface, improving adhesion, and prolonging drug contact time.13,14 This electrostatic attraction also enhances drug penetration by ensuring even distribution across the ocular surface, thus increasing the drug’s absorption into corneal and conjunctival tissues.13,14 These characteristics make cationic emulsions an effective vehicle for delivering therapeutic agents in the treatment of various ocular conditions, including dry eye disease and keratitis. Cationorm® (Santen Ltd, Japan) and Ikervis (0.1% cyclosporin A, Santen SAS, Evry, France) are examples.15–17,18,19 Ikervis contains 0.05 mg of CKC in 1 mL of emulsion, equivalent to a 0.005% solution. Both Cationorm and Ikervis have demonstrated efficacy in managing mild to moderate dry eye disease.
CKC exhibits similarities to BAK due to the presence of active quaternary ammonium components in both compounds. Previous studies have reported toxicity thresholds for BAK ranging from 0.0004% to 0.005%, while it is commonly used in ophthalmical formulations at concentrations between 0.02% and 0.04%.5–7 Studies in tissue culture and animal models have shown that BAK reduces cell survival in the corneal epithelium, conjunctival epithelium, trabecular meshwork cells, and ciliary epithelium.3,20–22 Additionally, it induces lymphocyte infiltration in conjunctival tissues and increases inflammatory markers.21,22 BAK interferes with mitochondrial ATP production in a concentration-dependent manner, even at low micromolar concentrations (IC50, 0.0002%). 8
The toxic effects of BAK on corneal endothelial cells have been previously reported. When cultured corneal endothelial cells were exposed for 48 h to antiglaucoma medications containing BAK diluted 10-fold, the cellular viability significantly decreased to 47–55.5%, compared with viability measured after exposure to the same medications without BAK (71–88.5%). 23 These antiglaucoma medications contain 0.015–0.005% of BAK. Inadvertent exposure of cornea endothelial cells to BAK-containing ophthalmical product resulted in significant toxicity.24,25
CKC has recently been introduced in the field of ophthalmology, and studies on its toxicity mechanism remain limited. Due to this lack of prior research, we hypothesized that CKC, as a quaternary ammonium compound, might induce toxicity in corneal cells through a mechanism similar to the well-documented toxic effects of BAK. Since BAK has been shown to increase ROS levels and cause mitochondrial damage, our study found that high concentrations of CKC similarly lead to elevated ROS production; however, it did not significantly alter mitochondrial distribution or morphology in surviving cells. Importantly, the absence of such abnormalities in surviving cells does not necessarily indicate that mitochondrial damage did not occur in cells that had already died.
Chronic ophthalmical disorders, such as glaucoma, necessitate prolonged daily administration of topical medications over several decades. Unlike corneal epithelial cells, which undergo complete turnover within approximately 2 weeks, corneal endothelial cells enter a postmitotic state after birth and experience a gradual, lifelong decline in cell density. 26 Considering the nonregenerative nature of corneal endothelial cells, a comprehensive assessment of the cytotoxic effects of ophthalmical drugs and their excipients on these cells is imperative for ensuring long-term ocular health.
The toxic concentration of CKC observed in corneal endothelial cells in this study closely parallels the findings from our previous investigations on corneal epithelial cells, suggesting a shared underlying mechanism of CKC-induced toxicity across these cell types. While a notable limitation of our experimental design is the inability to fully account for the fundamental biological differences between actively proliferating epithelial cells and postmitotic endothelial cells in vivo, our findings clearly indicate that toxic concentrations of CKC promote an increase in ROS and disrupt key cellular survival pathways.
It is important to note that less than 10% of an instilled eyedrop remains in the conjunctival fornix following administration, as approximately 90% is rapidly cleared from the ocular surface by reflex tearing and blinking. 27 Furthermore, for the small fraction that does remain, anatomical ocular barriers—including epithelial tight junctions, blood flow washout, and the cornea’s hydrophilic and hydrophobic properties—pose additional challenges that limit drug penetration through the cornea and sclera.28,29 Therefore, our 48–72 h continuous exposure model should be interpreted with caution when considering clinical relevance.
It is important to acknowledge certain limitations of this in vitro study. Methanol was used as a vehicle to dissolve CKC, and while its effects were considered in the toxicity assessment, its potential to amplify CKC’s toxic impact cannot be entirely ruled out. While cell culture models provide a controlled environment for investigating specific cellular mechanisms, they do not fully replicate the complex physiological environment of the in vivo eye. Factors such as tear film dynamics, blinking, corneal barrier function, and the presence of inflammatory and immune cells are absent in our experimental setup. These factors can influence the actual concentration of the preservative reaching the corneal endothelial layers, the duration of exposure, and the overall cellular response. Therefore, while our findings indicate a significant cytotoxic potential for CKC, in vivo studies are essential to fully ascertain its safety profile in a living system. Since this study primarily focused on short-term toxicity in cultured cells, further in vivo research is needed to assess the effects of prolonged CKC exposure over weeks or months. Another limitation is the use of an immortalized cell line to model corneal endothelial cells. In vitro, primary cultured corneal endothelial cells can proliferate only a few passages in vitro and are prone to mesenchymal transition, which complicates obtaining enough early-passage endothelial cells for research purposes. Therefore, when interpreting our results, it is important to consider the possibility that immortalized cell lines often show reduced sensitivity to toxicants. In addition, donor-to-donor variability and culture medium composition have a significant impact on growth rate, proliferation capacity, and morphology of the cells. 30 Primary cultured corneal endothelial cells may be more sensitive to toxicity compared with immortalized cell lines, and the possibility of endothelial–mesenchymal transition induced by toxicity cannot be ruled out.
Conclusions
In conclusion, this experiment has elucidated the potential concentration-dependent toxicity of CKC in HCEnCs. Various eye drops are formulated to penetrate corneal endothelial cells after being applied to the ocular surface, allowing the active components to reach intraocular target tissues at therapeutic concentrations. While regulating the concentration of the active component is crucial, the irreversible nature of corneal endothelial cells necessitates strict limitations on the inclusion of CKC as a preservative. This underscores the need for future eye drop formulations to ensure that CKC is maintained at a safe concentration level to minimize the risk of toxicity.
Authors’ Contributions
J.P.: Data curation, formal analysis, investigation, methodology, visualization, and writing—original draft. Y.S.: Formal analysis, investigation, methodology, visualization, and writing—original draft. C.Y.P.: Conceptualization, data curation, funding acquisition, formal analysis, investigation, validation, visualization, and writing—review and editing. All authors have read and agreed to the published version of the article.
Footnotes
Author Disclosure Statement
The authors declare no conflicts of interest. The authors have no conflict of interest in the materials presented herein.
Funding Information
The publication of this article was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (RS-2023-KH135936).
Data Availability Statement
The original data is available on request to the corresponding author.