
Abstract
The purpose is to review the role of the nitric oxide (NO)–soluble guanylate cyclase (sGC)–cyclic guanosine monophosphate (cGMP) signaling pathway in glaucoma, focusing on trabecular meshwork (TM) function, aqueous humor outflow, intraocular pressure (IOP), and ocular fibrosis. A structured narrative review of PubMed/MEDLINE, Embase, and Web of Science identified peer-reviewed studies published from January 2000 to March 2026 using terms including “soluble guanylate cyclase,” “sGC stimulator,” “sGC activator,” “cGMP,” “nitric oxide,” “glaucoma,” “trabecular meshwork,” “aqueous humor outflow,” and “ocular hypertension.” Experimental studies involving TM cells, Schlemm’s canal endothelial cells, ocular fibroblasts, anterior segment perfusion systems, animal models, and human clinical investigations were qualitatively reviewed. Evidence supports an important role for NO–sGC–cGMP signaling in regulating conventional aqueous humor outflow. sGC activation increases intracellular cGMP and promotes protein kinase G–mediated modulation of cytoskeletal organization and RhoA/Rho kinase signaling, reducing TM contractility and increasing outflow facility. cGMP signaling may also regulate extracellular matrix remodeling and transforming growth factor-β–associated fibrotic responses in ocular tissues. Preclinical studies demonstrate enhanced outflow facility and reduced IOP after pharmacologic pathway activation. Clinical evidence is strongest for NO-donating therapies such as latanoprostene bunod, whereas direct evidence for selective sGC stimulators and activators remains limited. The NO–sGC–cGMP pathway is a promising therapeutic target in glaucoma, particularly for trabecular outflow regulation and IOP control. Despite consistent mechanistic preclinical evidence, translational challenges remain, including drug delivery, tissue-specific pharmacodynamics, and limited clinical evaluation of direct sGC modulators. Further studies are needed to define the therapeutic role of selective sGC-targeted strategies in glaucoma management.
Keywords
Highlights
Introduction
Glaucoma is a leading cause of irreversible blindness worldwide and is characterized by progressive optic neuropathy commonly associated with elevated intraocular pressure (IOP).1,2 Increased resistance to aqueous humor drainage through the conventional outflow pathway, particularly at the level of the trabecular meshwork (TM) and Schlemm’s canal, is a major determinant of IOP elevation in primary open-angle glaucoma.3,4 Dynamic regulation of TM contractility, cytoskeletal organization, cell–matrix interactions, and extracellular matrix (ECM) turnover collectively influences outflow resistance and aqueous humor dynamics.3,4
Among the signaling pathways implicated in regulation of conventional outflow, the nitric oxide (NO)–soluble guanylate cyclase (sGC)–cyclic guanosine monophosphate (cGMP) pathway has emerged as an important modulator of ocular physiology.1,5 sGC is a cytosolic heterodimeric enzyme that functions as the principal intracellular receptor for NO. Binding of NO to the reduced ferrous (Fe2+) heme moiety of sGC activates the enzyme and catalyzes conversion of guanosine triphosphate (GTP) to cGMP.6,7 Increased intracellular cGMP subsequently activates downstream effectors, particularly cGMP-dependent protein kinase G (PKG), leading to modulation of cytoskeletal dynamics, inhibition of RhoA/Rho kinase signaling, and altered cellular contractility.6,7
In ocular tissues, NO–sGC–cGMP signaling has been implicated in regulation of TM biomechanics, Schlemm’s canal function, ocular vascular tone, and fibrotic remodeling. 1 Experimental studies have demonstrated expression and functional activity of sGC signaling components in TM cells, Schlemm’s canal endothelial cells, conjunctival fibroblasts, and corneal keratocytes.8,9 Activation of this pathway has been associated with relaxation of TM cells, increased conventional outflow facility, and reduction of IOP in experimental systems. 8 In addition, cGMP signaling may modulate transforming growth factor-β (TGF-β)-driven profibrotic responses, including myofibroblast differentiation and ECM deposition in ocular fibroblasts and corneal stromal cells. 9
The biologic activity of sGC is influenced by the redox state of its heme group. Under conditions of oxidative stress, the heme moiety may become oxidized or lost, rendering sGC less responsive to endogenous NO.10,11 This distinction has important pharmacologic implications and has led to the development of two mechanistically distinct classes of sGC-targeting agents: sGC stimulators and sGC activators.10–12 sGC stimulators enhance activity of the reduced heme-containing enzyme and act synergistically with endogenous NO, whereas sGC activators directly stimulate oxidized or heme-deficient sGC independently of NO availability.11,12 These pharmacologic strategies may be particularly relevant in glaucoma, where oxidative stress and impaired NO signaling have been implicated in outflow dysfunction.
Interest in therapeutic targeting of the NO–sGC–cGMP pathway in glaucoma has increased substantially over the past decade. The clearest clinical example is latanoprostene bunod, a NO–donating prostaglandin analog that combines enhancement of uveoscleral outflow with NO-mediated augmentation of conventional outflow through sGC activation. 13 Clinical studies have demonstrated effective IOP reduction and support the translational relevance of modulating this pathway in human glaucoma therapy.13,14 However, despite encouraging mechanistic and preclinical findings, direct clinical evidence for selective sGC stimulators and activators in ophthalmology remains limited. 15
This review summarizes current evidence regarding the role of the NO–sGC–cGMP signaling pathway in glaucoma, with emphasis on mechanisms of trabecular outflow regulation, preclinical evidence across experimental systems, clinical translation, and remaining translational challenges relevant to future therapeutic development.
Methods
Literature search strategy
This article is a structured narrative review evaluating the role of NO–sGC–cGMP signaling in glaucoma and ocular outflow regulation. Literature search was performed using PubMed/MEDLINE, Embase, and Web of Science databases to identify peer-reviewed studies published between January 2000 and March 2026. Search terms included combinations of “soluble guanylate cyclase,” “sGC stimulator,” “sGC activator,” “cGMP,” “nitric oxide signaling,” “glaucoma,” “trabecular meshwork,” “Schlemm’s canal,” “aqueous humor outflow,” “ocular hypertension,” “fibrosis,” “conjunctival fibroblasts,” and “corneal keratocytes.” Additional searches included specific pharmacologic compounds, including BAY 41-2272, BAY 58-2667 (cinaciguat), IWP-953, MGV354, and latanoprostene bunod. Reference lists of relevant articles were manually screened to identify additional studies.
Eligibility criteria and study classification
Studies were eligible for inclusion if they reported original experimental or clinical data related to: sGC signaling, cGMP-mediated mechanisms, pharmacologic sGC modulation, TM physiology, aqueous humor dynamics, ocular fibrosis, or IOP regulation. Included experimental systems comprised: TM cells, Schlemm’s canal endothelial cells, conjunctival fibroblasts, corneal keratocytes, anterior segment perfusion models, animal models of ocular hypertension or glaucoma, and human clinical studies. Review articles, conference abstracts without full datasets, and studies lacking mechanistic or translational relevance to ocular physiology were excluded from primary consideration, although selected reviews were used for contextual background. Studies were categorized according to level of evidence into mechanistic and molecular evidence, in vitro cellular studies, ex vivo and in vivo preclinical studies, and clinical investigations.
Data extraction and interpretation
Data extracted from eligible studies included experimental model, pharmacologic intervention, signaling pathways evaluated, cGMP-related mechanisms, TM or ECM effects, outflow facility measurements, IOP outcomes, and fibrosis-related endpoints. Because of substantial heterogeneity in study design, pharmacologic agents, outcome measures, and experimental systems, no quantitative meta-analysis was performed. Therefore, no Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) Flow Chart was included and findings were interpreted qualitatively with emphasis on mechanistic consistency, translational relevance, and differences across experimental levels.
Ethical considerations
This review is based exclusively on previously published studies and did not involve new experimentation in humans or animals. All included human investigations were conducted according to the Declaration of Helsinki, and all animal studies adhered to Association for Research in Vision and Ophthalmology (ARVO) and/or institutional guidelines for animal research as reported in the original publications.
Mechanisms of NO–sGC–cGMP Signaling in Ocular Outflow Regulation
sGC biology and redox regulation
sGC is the principal intracellular receptor for NO and a central mediator of cGMP signaling. 6 sGC is a heterodimeric cytosolic hemoprotein composed of α and β subunits, with the β1 subunit containing a prosthetic ferrous (Fe2+) heme moiety essential for NO binding and enzymatic activation. Binding of NO to the reduced ferrous heme induces conformational changes within the sGC heterodimer that enhance catalytic conversion of GTP to cGMP. Importantly, sGC exists in multiple redox-dependent functional states that determine its responsiveness to endogenous NO and pharmacologic modulation: the reduced Fe2+ heme-bound form, which is fully NO-sensitive and catalytically active; the oxidized Fe³+ heme state, which exhibits markedly diminished NO sensitivity; and the heme-deficient apo-sGC state, which is entirely NO-insensitive.
Pharmacologic targeting of sGC is therefore determined by the enzyme’s redox and heme-binding status, giving rise to two mechanistically distinct classes of modulators: sGC stimulators and sGC activators. sGC stimulators, including BAY 41–2272 and riociguat, selectively enhance activity of the reduced, heme-containing sGC (Fe2+ state). These agents stabilize the NO–heme–sGC complex, increase sensitivity to endogenous NO, and synergistically augment cGMP generation in the presence of physiologic NO signaling. Their efficacy consequently depends on preservation of the native ferrous heme moiety and at least partial endogenous NO bioavailability.
In contrast, sGC activators, such as cinaciguat (BAY 58-2667), preferentially target oxidized (Fe³+) or heme-deficient apo-sGC conformations by binding directly within the vacant or destabilized heme pocket, thereby restoring enzymatic activity independently of NO. These compounds bypass the requirement for endogenous NO signaling and re-establish cGMP production under conditions of oxidative stress, remaining active even when canonical NO–sGC signaling is severely impaired.10–12
This distinction is particularly relevant in glaucomatous tissues, where oxidative heme loss and sGC oxidation may limit the efficacy of NO-dependent therapies while rendering oxidized or apo-sGC a viable target for activator-based interventions FIG. 1. Accordingly, dysregulation of the NO–sGC–cGMP axis in glaucoma should not be viewed solely as a consequence of reduced NO bioavailability, but also as a structural and redox-dependent enzymopathy characterized by heme destabilization and loss of sGC functional integrity.6,7 Oxidative stress may oxidize the heme moiety or induce heme loss, producing an NO-insensitive enzyme state associated with impaired cGMP generation.10,11 This mechanism may contribute to TM dysfunction and increased aqueous outflow resistance linked to oxidative stress, endothelial dysfunction, and reduced NO bioavailability.5,16,17
Recognition of these redox-dependent signaling states has driven development of two complementary pharmacologic strategies FIG. 1: sGC stimulators, which enhance signaling through reduced heme-containing sGC in synergy with endogenous NO, and sGC activators, which directly activate oxidized or heme-deficient sGC independently of NO availability.10–12 These approaches may therefore retain therapeutic efficacy under oxidative conditions associated with impaired endogenous NO signaling. 18 Increased intracellular cGMP subsequently activates downstream effectors including cGMP-dependent PKG, cyclic nucleotide-gated ion channels, and cGMP-regulated phosphodiesterases.6,7
Guanylyl cyclases comprise two major biological families: sGCs (sGCs) and membrane-bound particulate guanylate cyclases (pGCs). Unlike sGC, which is activated by NO, pGC receptors are stimulated by natriuretic peptides including atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP) (see section 3.2).19–21 Although both pathways generate cGMP, current pharmacologic approaches relevant to glaucoma predominantly target NO-dependent sGC signaling.
Particulate guanylyl cyclases (pGCs)
In contrast to sGC, particulate guanylyl cyclases (pGCs) are transmembrane receptors activated by extracellular peptide ligands rather than gaseous signaling molecules. These enzymes include several isoforms, most notably guanylyl cyclase-A (GC-A) and guanylyl cyclase-B (GC-B), which serve as receptors for atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP).20,21 Upon ligand binding, pGC receptors undergo conformational activation of their intracellular catalytic domains, leading to cGMP production. Although pGC-generated cGMP activates many of the same downstream effectors as sGC-derived cGMP, including PKG, the spatial and temporal characteristics of signaling differ because of membrane localization and receptor-specific regulatory mechanisms. 20
In ocular tissues, natriuretic peptide signaling has been implicated in regulation of aqueous humor dynamics, ocular blood flow, and IOP. Experimental studies have shown that ANP and CNP can influence outflow facility and ciliary body function, suggesting a modulatory role of pGC signaling in ocular physiology. 21 However, compared with sGC, pharmacologic targeting of pGC pathways in ophthalmology remains relatively underdeveloped and is not currently a major focus of therapeutic intervention.
Although both sGC and pGC pathways generate cGMP, they differ fundamentally in ligand activation, cellular localization, and redox sensitivity. In ophthalmology, current therapeutic strategies targeting cGMP signaling predominantly act through the NO–sGC axis rather than natriuretic peptide–dependent pGC pathways. Accordingly, this review focuses primarily on sGC signaling because of its central role in TM regulation, IOP homeostasis, and oxidative stress-associated ocular pathology.
cGMP signaling and cytoskeletal regulation
The principal downstream effector of cGMP signaling is PKG, which regulates pathways involved in cytoskeletal organization, smooth muscle tone, cellular adhesion, and contractility.6,7 In TM cells, PKG activation suppresses RhoA/Rho-associated protein kinase (ROCK) signaling, resulting in reduced myosin light chain phosphorylation, disruption of actin stress fibers, decreased focal adhesion formation, and reduced cellular contractility.22,23 Collectively, these effects decrease TM stiffness and enhance conventional aqueous humor outflow. Experimental studies have demonstrated substantial reductions in actin stress fiber formation and TM cellular stiffness following pharmacologic activation of cGMP signaling pathways.24,25 These biomechanical alterations are mechanistically consistent with enhanced aqueous humor drainage through Schlemm’s canal and associated outflow tissues.
Beyond cytoskeletal regulation, cGMP signaling functions as a spatially compartmentalized intracellular signaling network. Localized cGMP pools, organized through scaffold proteins and phosphodiesterase (PDE)-defined microdomains, permit selective activation of downstream effectors despite use of a shared second messenger. 26 This compartmentalization may be particularly important in ocular tissues, where regulation of TM contractility, Schlemm’s canal endothelial permeability, fibroblast activation, and mechanotransduction pathways may depend on microdomain-specific rather than global intracellular cGMP signaling.
cGMP–PKG signaling also interacts with multiple pathways involved in cellular phenotype regulation and mechanobiology, including TGF-β/SMAD, mitogen-activated protein kinase (MAPK/ERK), phosphoinositide 3-kinase (PI3K)/AKT, and RhoA/ROCK signaling networks. Through these interactions, cGMP signaling serves as an integrative regulatory node linking mechanical, inflammatory, and growth factor-mediated signaling within TM tissues.
The magnitude and duration of cGMP signaling are tightly regulated by PDEs, which hydrolyze cGMP to inactive 5′-GMP.27,28 Among these, PDE5 plays a major role in ocular cGMP degradation, although PDE6 and PDE9 may also contribute to cGMP turnover in ocular tissues.29,30 PDE expression and activity regulate the amplitude and duration of PKG activation and may substantially influence tissue responsiveness to pharmacologic sGC modulation. Variability in phosphodiesterase activity, endogenous NO bioavailability, and oxidative stress burden may therefore contribute to differences in signaling responsiveness across experimental systems and disease states. Mechanistically, both ROCK inhibitors and sGC pathway modulators influence TM cytoskeletal dynamics and outflow resistance; however, ROCK inhibitors directly inhibit Rho-associated kinase activity, whereas sGC signaling modulates these pathways indirectly through cGMP-dependent PKG activation upstream of cytoskeletal remodeling cascades.31–35
ECM remodeling and fibrotic signaling
In addition to regulating TM contractility, NO–sGC–cGMP signaling influences ECM turnover and fibrosis-related pathways within ocular tissues. Activation of PKG has been associated with inhibition of TGF-β-mediated profibrotic signaling, including suppression of SMAD-dependent transcriptional programs involved in myofibroblast differentiation and matrix deposition.9,26,27 PKG signaling additionally interacts with RhoA/ROCK, MAPK/ERK, and PI3K/AKT pathways, positioning cGMP signaling as an integrated regulator of fibroblast phenotype, mechanotransduction, and tissue remodeling dynamics.
Experimental studies in conjunctival fibroblasts and corneal keratocytes have demonstrated reduced expression of α-smooth muscle actin (α-SMA), collagen type I, fibronectin, and other fibrosis-associated markers following pharmacologic stimulation of sGC signaling pathways.9,26 In ocular fibroblast systems, BAY 41-2272 has been associated with substantial reductions in α-SMA and collagen type I expression.9,26 These effects are accompanied by increased matrix metalloproteinase activity and reduced expression of tissue inhibitors of metalloproteinases, favoring ECM turnover over matrix accumulation.26,27
Collectively, these signaling effects shift ocular tissues toward a matrix-degrading, compliance-enhancing phenotype that may be relevant to both TM dysfunction and postoperative fibrosis FIG. 2. Thus, NO–sGC–cGMP signaling may contribute not only to acute regulation of aqueous humor outflow resistance through cytoskeletal relaxation, but also to longer-term tissue remodeling processes involved in glaucomatous dysfunction and surgical wound healing Table 1. Although these findings are mechanistically consistent across several experimental systems, the relative contribution of ECM remodeling versus acute cytoskeletal relaxation to sustained IOP reduction remains incompletely defined.
Mechanistic and Experimental Evidence for NO–sGC–cGMP Modulation in Ocular Outflow Pathways
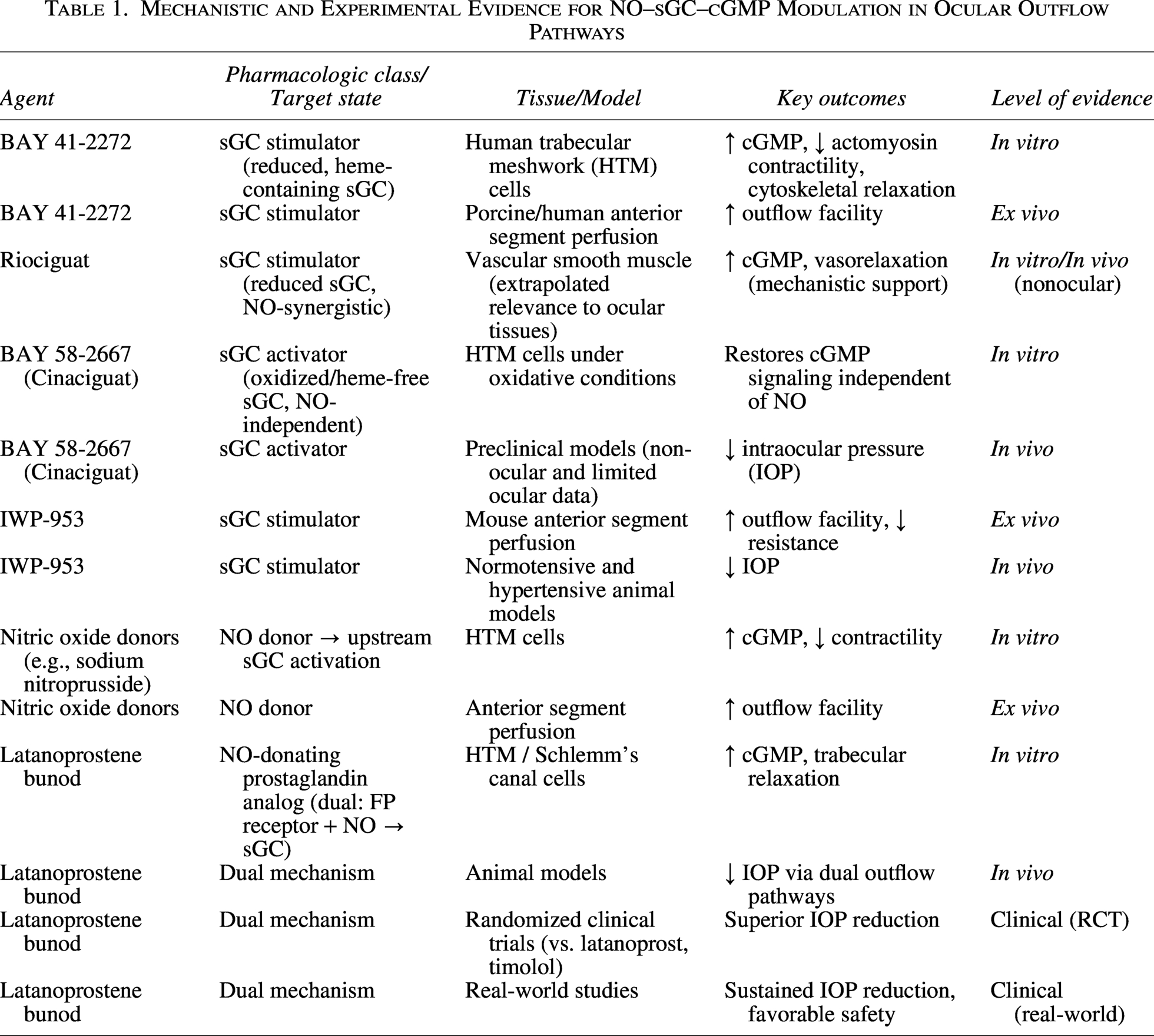
Preclinical and Clinical Evidence
In vitro evidence
In cultured human TM (HTM) cells, activation of the NO–sGC–cGMP pathway consistently reduces cellular contractility and alters cytoskeletal organization. Dismuke et al. demonstrated concentration-dependent relaxation responses following exposure to NO donors, accompanied by reductions in actin stress fibers and actomyosin-mediated contractility. 35 Similar findings have been observed with direct sGC stimulation using BAY 41-2272 and related compounds, which increase intracellular cGMP levels and suppress RhoA/ROCK signaling pathways.8,22,35
Additional studies suggest that sGC signaling influences TM biomechanics through modulation of cellular stiffness, focal adhesion dynamics, and integrin-mediated cytoskeletal organization.24,25 These molecular and cellular effects are mechanistically consistent with increased aqueous humor outflow through the conventional drainage pathway.
NO-mediated signaling has also been implicated in regulation of Schlemm’s canal endothelial permeability and cellular morphology, although available evidence remains more limited than for TM systems.1,3 Experimental studies suggest that modulation of endothelial barrier properties within Schlemm’s canal may contribute to regulation of outflow resistance and pressure-dependent aqueous humor drainage.
Antifibrotic effects of cGMP signaling have additionally been investigated in ocular stromal cells. Fioretto et al. demonstrated that pharmacologic stimulation of sGC with BAY 41-2272 attenuated TGF-β1-induced profibrotic activation in human conjunctival fibroblasts, including reductions in α-SMA expression and ECM-related gene transcription. 26 Similar findings were reported in human corneal keratocytes, where BAY 41-2272 reduced TGF-β1-induced myofibroblast differentiation and fibrosis-associated marker expression. 9 Collectively, available in vitro evidence supports a mechanistic role for NO–sGC–cGMP signaling in regulation of TM contractility, cytoskeletal dynamics, ECM turnover, and fibrosis-related cellular responses relevant to glaucoma pathophysiology.
Ex vivo and in vivo preclinical evidence
Ex vivo anterior segment perfusion systems provide direct functional assessment of aqueous humor outflow facility under controlled experimental conditions. In murine anterior segment perfusion models (isolated enucleated mouse anterior segment perfusion preparations), Ge et al. reported that the sGC stimulator IWP-953 significantly increased conventional outflow facility compared with vehicle-treated controls. 8 NO donors have similarly demonstrated increases in outflow facility that vary according to species and experimental conditions.36,37 These findings support the concept that molecular and cellular effects observed in vitro translate into tissue-level modulation of aqueous humor dynamics.
In vivo animal studies further support the physiologic relevance of NO–sGC–cGMP signaling in ocular pressure regulation. Pharmacologic activation of sGC has been associated with reduction of IOP in rabbit, porcine, canine, and nonhuman primate models.38–40 Prasanna et al. reported that topical administration of the selective sGC activator MGV354 produced significant reductions in IOP in normotensive and ocular hypertensive animal models following topical ocular dosing. 39 Similarly, NO–donating compounds such as NCX 667 reduced IOP in rabbits, dogs, and nonhuman primates while enhancing outflow responses in TM/Schlemm’s canal constructs exposed to TGF-β2. 40
Although preclinical findings have been mechanistically consistent across multiple experimental systems, interpretation should consider important limitations. Cellular and ex vivo systems provide important mechanistic insight but may not fully reproduce the complexity of IOP regulation in vivo. Likewise, animal models differ substantially from human glaucoma in anatomy, oxidative stress biology, aqueous humor dynamics, and outflow physiology. Consequently, translation from mechanistic findings to clinical efficacy should not be assumed to be linear.
Clinical evidence and translational relevance
The strongest clinical evidence supporting therapeutic modulation of the NO–sGC–cGMP pathway in glaucoma derives from NO–donating therapies, particularly latanoprostene bunod.13,41 Latanoprostene bunod is metabolized into latanoprost acid and a NO–donating moiety. Latanoprost acid enhances uveoscleral outflow through prostaglandin F receptor activation, whereas NO activates sGC signaling within TM and Schlemm’s canal tissues, increasing cGMP production and facilitating conventional aqueous humor outflow.41,42
Phase III APOLLO and LUNAR clinical trials demonstrated greater IOP reduction with latanoprostene bunod compared with timolol in patients with open-angle glaucoma or ocular hypertension.43,44 Mean IOP reductions from baseline ranged approximately from 7 to 9 mmHg, with additional IOP lowering beyond that achieved with latanoprost alone.41–43 Meta-analytic studies have similarly demonstrated superior or noninferior efficacy compared with established glaucoma therapies.13,45 Long-term data from the JUPITER study demonstrated sustained efficacy and acceptable tolerability over 12 months of treatment. 46 Real-world observational studies have additionally reported durable IOP reduction and safety profiles generally comparable to those of other prostaglandin analog therapies.14,47 In contrast, direct clinical evidence for selective sGC stimulators and activators remains limited. Early-phase clinical evaluation of MGV354 demonstrated acceptable ocular safety but relatively modest IOP-lowering efficacy. 15 Overall, currently available evidence supports biological and translational relevance of NO–sGC–cGMP signaling in glaucoma, while emphasizing the need for additional clinical studies evaluating selective sGC-targeted therapies in human disease (Table 2).
Quantitative Functional Outcomes of NO–sGC–cGMP Pathway Modulation Relevant to Intraocular Pressure Regulation
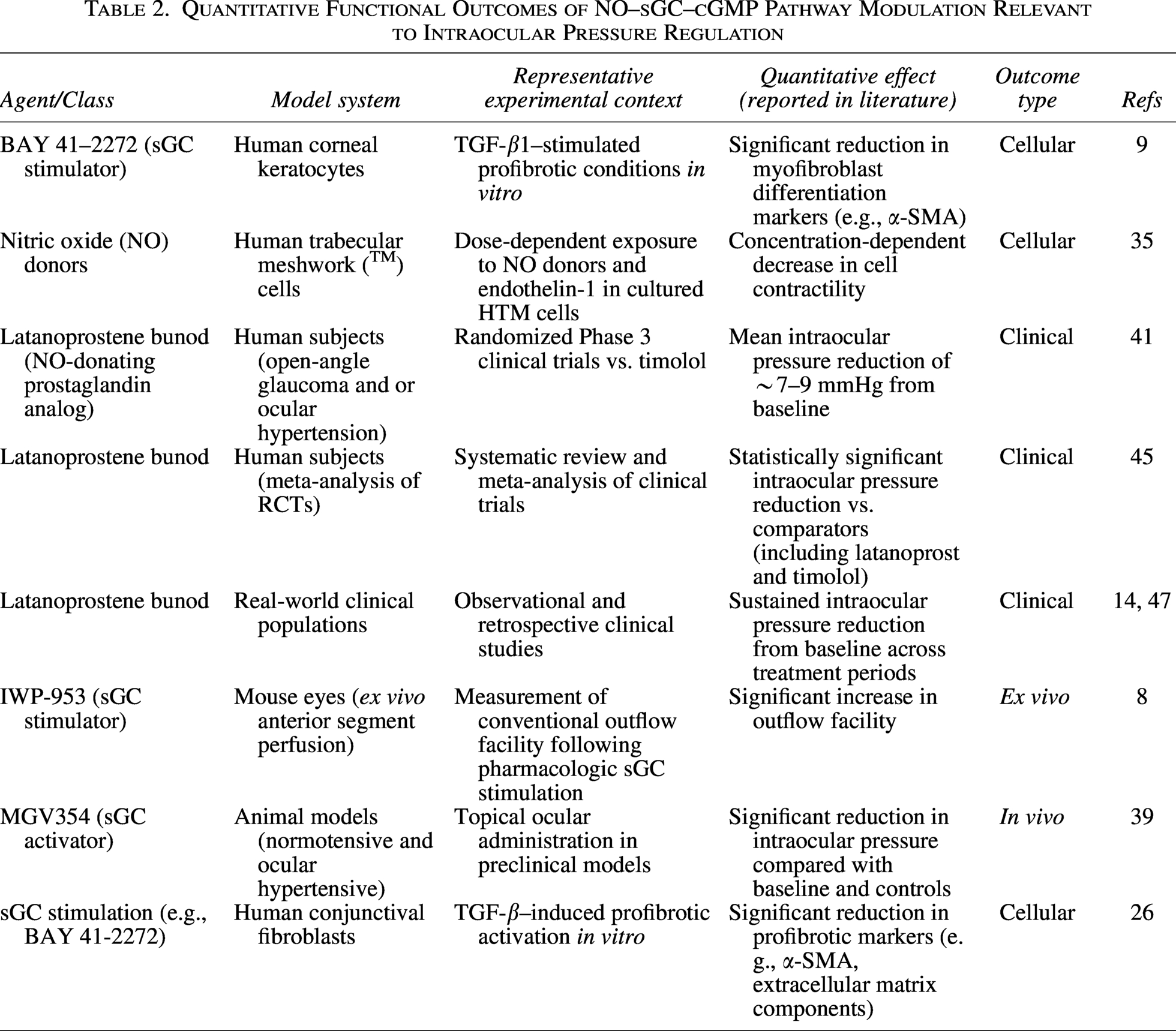
Quantitative pharmacodynamics of NO–sGC–cGMP modulation
Pharmacologic activation of sGC produces measurable, concentration-dependent increases in intracellular cGMP. In biochemical and vascular smooth muscle systems, sGC stimulators such as BAY 41–2272 demonstrate half-maximal effective concentrations (EC50) in the submicromolar to low micromolar range (∼0.1–1 µM), with maximal cGMP elevations that vary according to the experimental system. 48 Similarly, riociguat exhibits EC50 values near 0.1 µM, in the presence of NO, can amplify cGMP production relative to baseline, reflecting synergistic activation of the reduced, heme-containing enzyme.49,50
In ocular-relevant cellular systems, including TM cells, activation of the NO–sGC pathway results in quantifiable reductions in actomyosin contractility. Experimental studies report decreases in stress fiber formation and reductions in cellular stiffness, as measured by cytoskeletal imaging and atomic force microscopy. These changes correspond to functional modulation of TM biomechanics and are consistent with increased aqueous humor outflow observed in tissue-level systems.24,25
Ex vivo anterior segment perfusion models provide direct quantitative evidence of outflow modulation. Pharmacologic stimulation of sGC with compounds such as IWP-953 has been shown to substantially increase conventional outflow facility over baseline in murine models.8,36 NO donors, including sodium nitroprusside, produce comparable increases in outflow facility, depending on species and baseline outflow resistance. 37
In vivo preclinical models further demonstrate physiologic relevance. Topical administration of sGC activators, including cinaciguat (BAY 58-2667), results in moderate IOP reductions in animal models, particularly under conditions associated with oxidative stress. These findings are supported by broader pharmacologic evidence demonstrating that sGC activators retain efficacy in oxidized or heme-free enzyme states, restoring cGMP signaling where responsiveness to NO or sGC stimulators is diminished.39,51,52
The magnitude and duration of cGMP signaling are further modulated by PDEs. Inhibition of PDE5, a cGMP-selective phosphodiesterase expressed in vascular and ocular tissues, prolongs intracellular cGMP signaling and enhances downstream signaling responses in smooth muscle, demonstrating that degradation kinetics are a key determinant of pharmacodynamic effect size.53,54
However, an important limitation of the current evidence base is the inconsistent reporting of quantitative pharmacodynamic parameters across ocular experimental systems. Although multiple studies demonstrate biologic activity of sGC modulators, concentration–response relationships, maximal efficacy estimates, and standardized target-engagement measures are variably reported and frequently assessed under noncomparable experimental conditions. This limits formal cross-study pharmacologic comparison and constrains precise assessment of relative potency, efficacy, and translational therapeutic margins across distinct sGC-targeted compounds.
Translational Challenges and Future Directions
Although substantial mechanistic and preclinical evidence supports a role for NO–sGC–cGMP signaling in regulation of trabecular outflow and IOP, significant translational limitations remain. A key challenge is the divergence between robust cellular and animal model data and the comparatively limited clinical efficacy data for direct sGC-targeted therapies. While NO–donating compounds such as latanoprostene bunod have demonstrated clinically meaningful IOP reduction, selective sGC stimulators and activators have not yet shown comparable efficacy in human studies FIG. 3.15,41,43
Several factors likely contribute to this translational gap. Ocular drug delivery constraints—including precorneal loss, limited corneal penetration, and rapid clearance—restrict effective exposure of TM and Schlemm’s canal tissues. In parallel, sGC activity is strongly influenced by local redox state and endogenous NO bioavailability, both of which vary across disease stages and patient populations.10–12,16,17 This heterogeneity may substantially affect responsiveness to specific sGC modulators.
Additional limitations arise from the heterogeneity of experimental systems. In vitro TM cell studies and ex vivo perfusion models incompletely replicate the integrated physiology of aqueous humor dynamics in vivo. 55 Likewise, animal models of ocular hypertension differ from human glaucoma in anatomy, outflow pathway biology, oxidative stress responses, and the presence or absence of chronic neurodegenerative components.56,57 These differences complicate direct extrapolation of preclinical efficacy to clinical outcomes.
The spatial and biochemical regulation of cGMP signaling within ocular tissues remains incompletely defined. Compartmentalization of cGMP by scaffold proteins and PDEs, together with microenvironmental influences, introduces variability in signaling dynamics.28–30 Quantitative pharmacodynamic characterization of ocular sGC modulation is also limited, with most studies using heterogeneous models, restricted dose ranges, and variable baseline NO conditions, thereby limiting robust comparison of EC50, maximal efficacy, and duration of response across systems. As a result, translation from mechanistic signaling effects to clinical IOP reduction cannot be assumed to be linear.
Mechanistically, the translational gap is further influenced by ocular pharmacokinetic constraints, redox-dependent variability in sGC activity, PDE-mediated cGMP degradation, and species-specific differences in outflow pathway regulation. Incomplete target engagement in clinical settings—particularly uncertain drug penetration into TM microdomains and limited evidence of sustained intraocular cGMP elevation in humans—may further reduce therapeutic impact.
Future directions should prioritize: (1) clinical evaluation of selective sGC stimulators and activators in glaucoma; (2) improved characterization of sGC redox states in human glaucomatous tissues to enable patient stratification; (3) development of targeted ocular delivery systems to enhance local bioavailability; and (4) exploration of combined modulation of sGC and PDE pathways to sustain cGMP signaling within the conventional outflow pathway. Overall, the NO–sGC–cGMP axis remains a biologically compelling therapeutic target, but requires more rigorous translational validation and clinically grounded pharmacodynamic characterization.
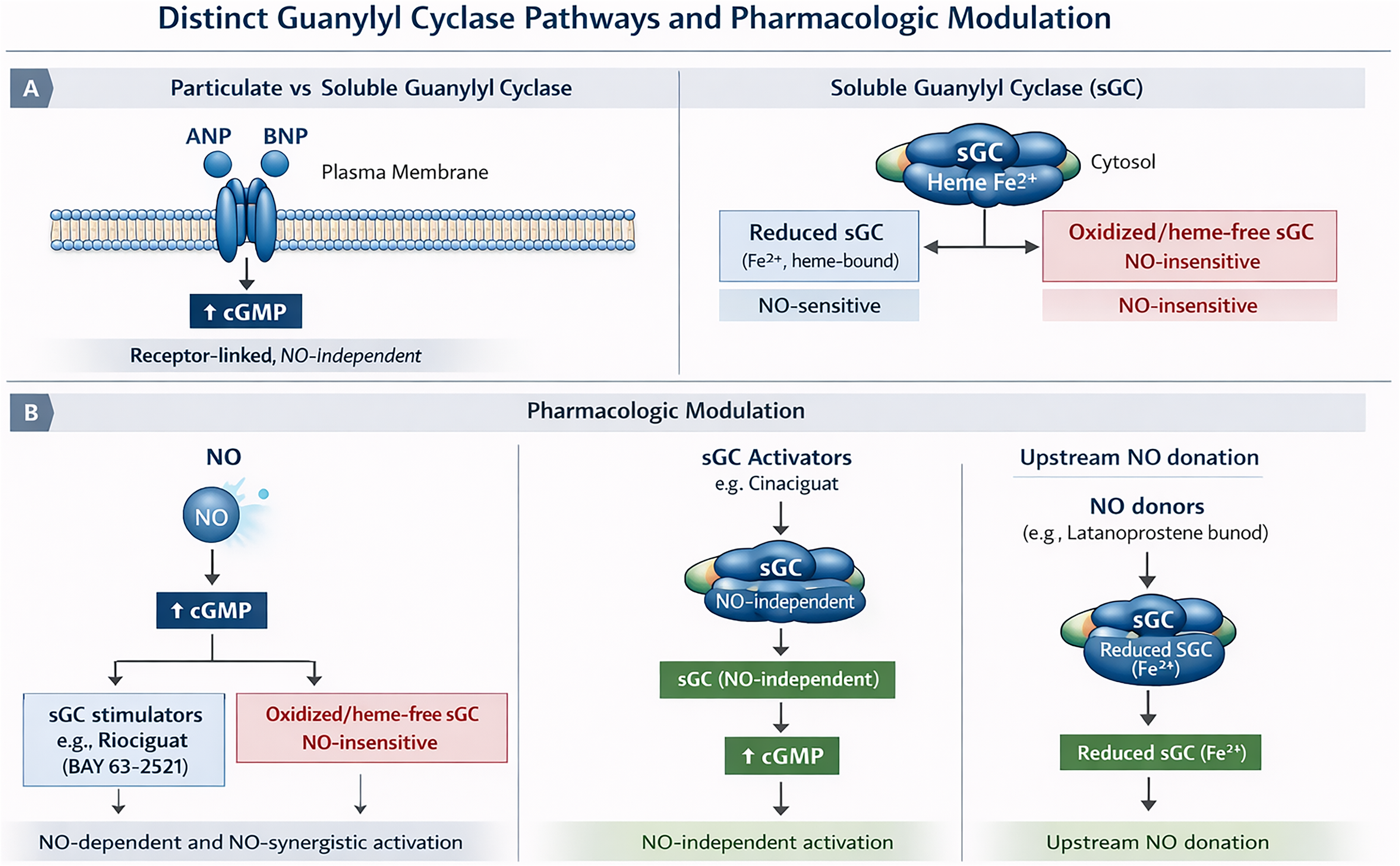
Soluble guanylate cyclase signaling and pharmacologic modulation. Nitric oxide activates reduced heme-containing soluble guanylate cyclase (sGC), increasing cyclic guanosine monophosphate (cGMP) production. sGC stimulators enhance signaling through reduced sGC, whereas sGC activators directly stimulate oxidized or heme-deficient sGC independently of nitric oxide.
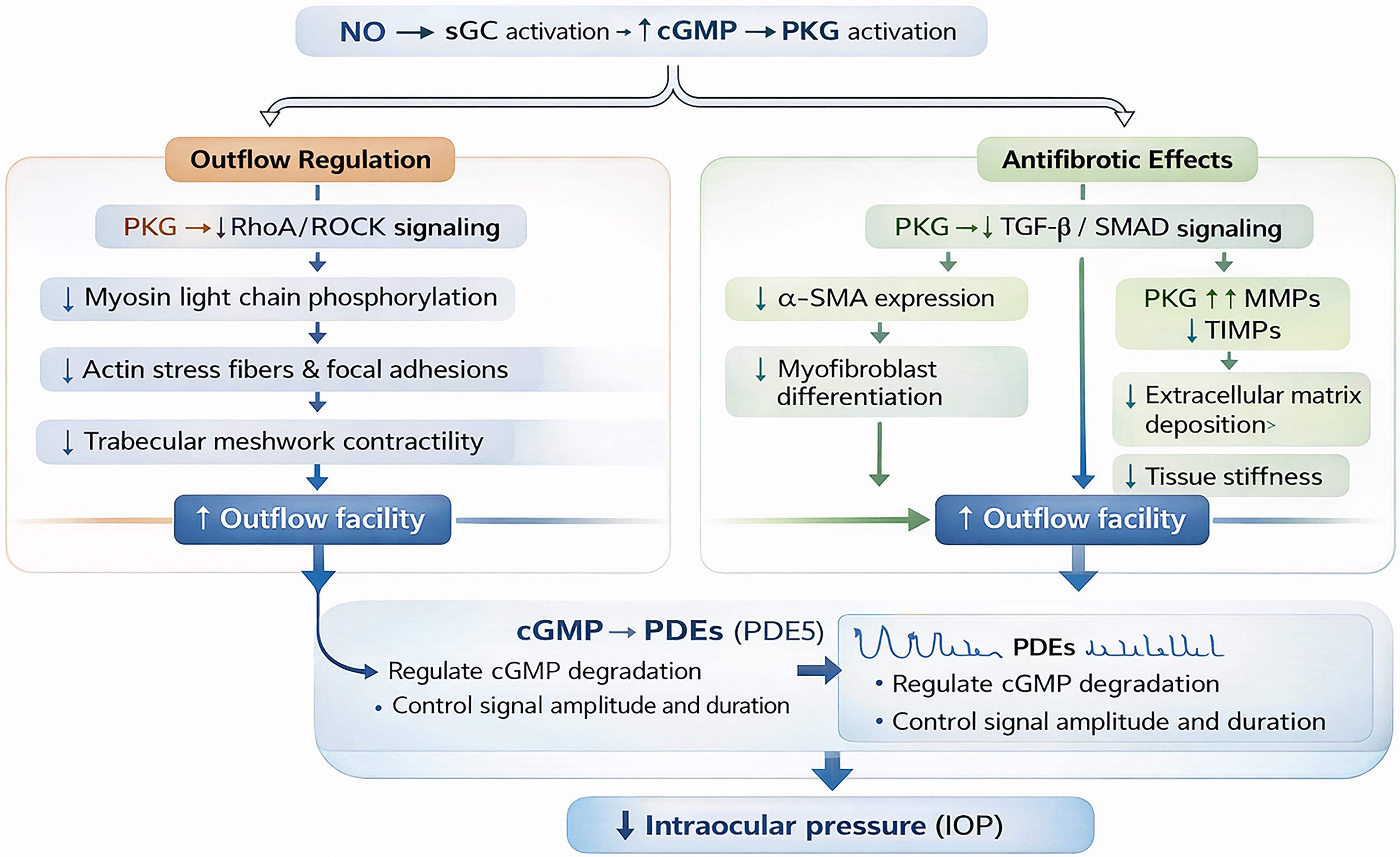
Downstream effects of NO–sGC–cGMP signaling in ocular outflow tissues. Activation of cGMP-dependent protein kinase (PKG) modulates cytoskeletal organization, inhibits RhoA/Rho kinase signaling, and reduces trabecular meshwork contractility. Additional effects on extracellular matrix turnover and fibrosis-related signaling may contribute to altered outflow resistance.
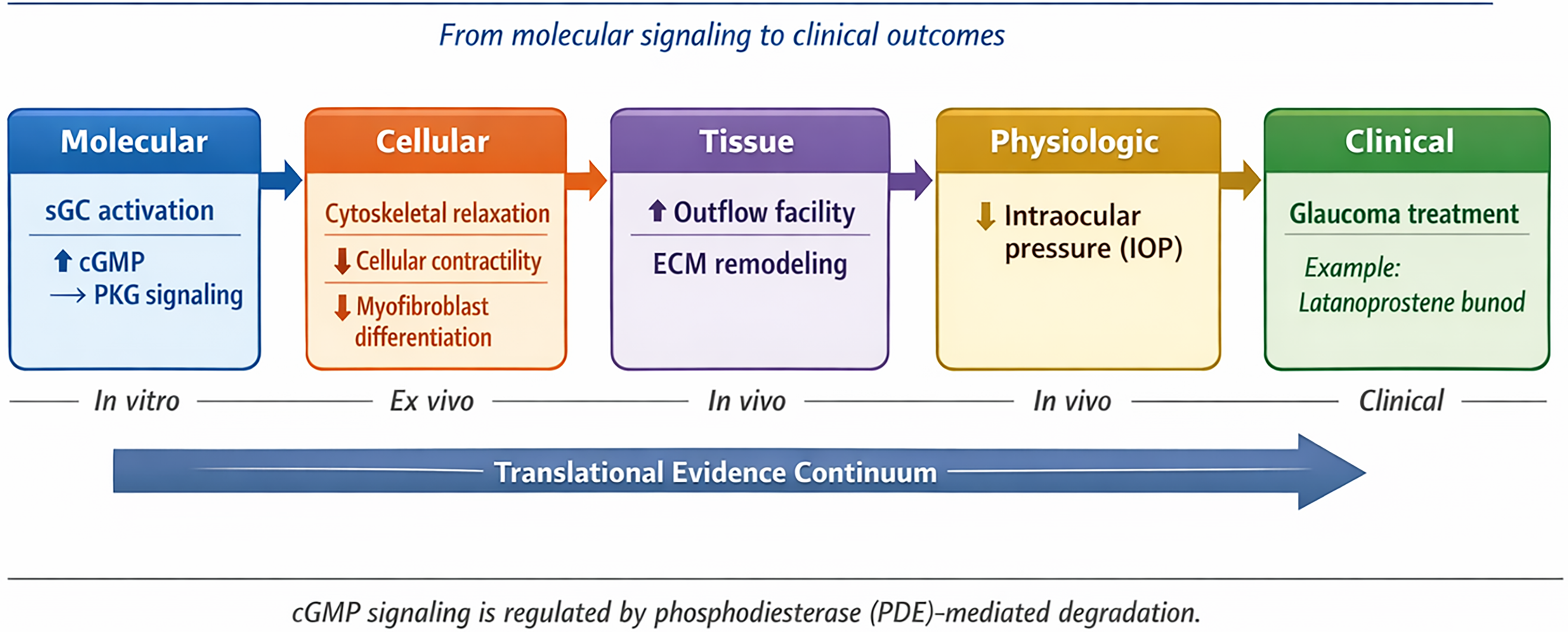
Translational continuum of NO–sGC–cGMP pathway modulation in glaucoma. Molecular activation of soluble guanylate cyclase influences cellular contractility, extracellular matrix remodeling, aqueous humor outflow, and intraocular pressure regulation across experimental and clinical systems.
Conclusions
The NO–sGC–cGMP pathway plays a central role in the regulation of TM function, aqueous humor outflow, and IOP homeostasis. Experimental studies have shown that activation of sGC enhances intracellular cGMP signaling and modulates cytoskeletal organization, RhoA/Rho kinase activity, and ECM remodeling within ocular outflow tissues. Collectively, these molecular and cellular effects are associated with increased conventional outflow facility and reduced IOP across multiple preclinical models. Beyond its effects on aqueous humor dynamics, cGMP signaling may also influence fibrosis-related pathways in ocular fibroblasts and stromal tissues, suggesting a potential role in postoperative scarring and tissue remodeling. To date, clinical translation of this pathway has been most clearly demonstrated by NO–donating therapies such as Latanoprostene bunod, which lower IOP through combined enhancement of uveoscleral and trabecular outflow. However, direct clinical evidence supporting the use of selective sGC stimulators and activators in ophthalmology remains limited.
This review has several limitations. As a narrative review, it is inherently subject to the limitations of non-quantitative evidence synthesis, including potential publication bias, heterogeneity among experimental models, and limited comparability across pharmacologic agents and outcome measures. Furthermore, much of the currently available evidence is derived from in vitro and ex vivo studies that may not fully replicate the physiologic complexity of human ocular tissues. Direct clinical evidence evaluating selective sGC stimulators and activators in ophthalmology also remains scarce, warranting caution when extrapolating mechanistic and preclinical findings to therapeutic efficacy in human glaucoma. Despite promising mechanistic and translational data, several challenges remain, including optimization of ocular drug delivery, clarification of tissue-specific pharmacodynamics, and a more comprehensive understanding of sGC redox regulation in glaucomatous tissues. Accordingly, further translational and clinical investigations are needed to better define the therapeutic potential of selective sGC-targeted therapies in glaucoma management.
Footnotes
Author’s Contributions
The corresponding author is the sole contributor, responsible for conception and design, literature review, formulation of research questions and hypotheses, data extraction and synthesis, drafting and revising the article, preparation of figures and tables, and final approval of the submitted version.
Availability of Data and Materials
No new patient data were generated or analyzed in this work.
Ethics Approval and Consent to Participate
Not applicable; no animal or patient experiments were conducted.
Intellectual Property Statement
All original ideas, interpretations, and conceptual contributions in this work are the sole intellectual property of the corresponding author. Any innovative notions or statements introduced in this review, or derived from its content, are exclusively attributed to the author.
AI—Declaration
No AI tools were used in the preparation of this work, except for the generation of figures, which were created with the assistance of AI software using the article text as input.
Author Disclosure Statement
The author declares no competing interests. This research was conducted independently and received no external funding.
Funding Information
No
additional external funding was received for this work.