
Abstract
Background
A simple yet powerful tool for providing for rapid gene identification in the clinic would be the combination of isothermal gene amplification with electronic micro-chip analysis. This is a first report of such a union of these technologies.
Methods
The first assay demonstrates discrimination between four bacterial pathogens. For this, one portion of the bacterial 16S rRNA gene encompassing a microheterogeneous region was isothermally amplified using Strand Displacement Amplification (SDA). Type identification was then made by “sandwich” assay format either using selective electronic hybridization of amplicons to sequence-specific capture oligonucleotides and a universal, fluorescently labeled reporter oligonucleotide, or, alternatively, sequence-specific reporters and a universal capture oligonucleotide. The second assay tested for the presence or absence of the Factor V Leiden point mutation using DNA obtained from 18 patients in a blind assay. For this, allele-specific SDA was developed. Following amplification using a sense-biotinylated primer and either the corresponding antisense wild type or mutant primer, multiple patient amplicons were targeted to specified locations on the microarray and visualized using a fluorescently labeled reporter oligonucleotide. Positive signals were scored as greater than or equal to two times the background.
Results
Bacterial type-specific signals were between 3- to 10-fold greater than nonspecific in both assay formats. Using allele-specific SDA, 100% agreement was observed between PAGE analysis, microarray results, and clinical diagnosis in Factor V mutation analysis.
Conclusions
We demonstrated two model clinical assays combining amplified materials and microelectronic arrays, one potentially suitable for pathogen screening and the other for a deleterious genetic mutation.
Introduction
Among evolving diagnostic tools, DNA analysis is steadily gaining importance. This is true both for the rapid identification of pathological organisms and for the detection of genetic predispositions or somatic mutations. To address these needs, the field of DNA diagnostics has moved toward gene amplification followed by analysis. Hybridization analysis offers many advantages over other methods of DNA identification, such as comparative size or relative migration determination, by providing insight into the specific nucleotide sequence being investigated. Specifically, with hybridization, spatial position of sequence-specific probes and signal intensity provide the basis for analyzing complex genetic mixtures for sequence-specific characteristics.
Under appropriate circumstances, the rapidity and sensitivity of hybridization analysis may be aided by miniaturization. Such miniaturized devices have been successfully developed for expression monitoring1–5 and analysis of single nucleotide polymorphisms (SNPs)6,7 within an amplified product (amplicon). Thus, these devices offer the potential for combining the specificity of hybridization with the speed and sensitivity of microchip technology. However, these microdevices can analyze multiple amplicons simultaneously only if hybridization conditions for each amplicon are compatible. This may be partially compensated for by careful capture oligonucleotide design, by the use of very long captures (eg, cDNA for expression monitoring),1,3 or by extensive redundancy and overlap of shorter capture oligonucleotide sequences. Taken together, these considerations have imposed limitations on the use of most microchip devices. For example, high levels of redundancy using short oligonucleotide captures require large arrays and complex informatics programs for interpretation of results. However, there may still be sequence-specific regions that remain difficult to analyze. Alternatively, the use of long capture oligonucleotides permits a uniformly elevated hybridization temperature to be used. However, long oligonucleotides are usually incompatible with single base pair mismatch analysis.
A different class of microscale device for the analysis of oligonucleotides is that of the electronically controlled microelectrode array.8,9 In contrast to the passive and uniform hybridization environment of most other microscale devices, these electronic chip-based devices offer the ability to actively transport DNA to, and hybridize at, discreet locations on the microelectrode array surface.8,9 In concept, these active microarrays circumvent many of the limitations encountered by other microdevices. These active microchip arrays overcome the size dependency of capture oligonucleotides and/or complexity requirements of passive microdevices. Also, these microchips allow multiple independent analyses upon the same open microarray surface by selectively and independently targeting different DNA samples to various microelectrode locations. In other words, they allow parallel multiple sample processing on an open array.
To provide material for analysis, we used the technique of Strand Displacement Amplification (SDA).10 This is a rapid, isothermal amplification technique that is easily adapted to chips and microinstrumentation. In brief, the amplification process uses a restriction endonuclease nicking step followed by 5' to 3' displacement of the residual hybridized sequence. This displacement is caused by polymerase activity initiated at the nick site during sequence extension, thereby avoiding the need for temperature ramping for denaturation of hybridized strands. In general, this simplifies associated controls and electronics. The ultimate goal is to combine this technology with electronic chip technology to provide for a simple and compact platform on which to perform genetic diagnostic analysis in the clinic.
Broadly stated, this diagnostic analysis comprises two related but distinct forms of DNA analysis. The first can be described as “one patient, many possible pathogens or genes” for a patient-oriented program and the second can be thought of as “one gene, many patients” for a screening program. To demonstrate the capabilities of isothermal amplification combined with electronic targeting to meet these dual requirements, we present two prototypic assays: one of amplified bacterial test samples being simultaneously analyzed for one of several pathogenic bacteria commonly found in infectious diarrhea outbreaks and, separately, of multiple patient DNA samples being simultaneously analyzed for a common genetic mutation, which predisposes to significant morbidity from deep venous thrombosis.11,12 In order to provide test material for the latter analysis, we developed allele-specific SDA.
Methods
Materials
Deoxynucleoside 5'-triphosphates (dGTP, dATP, dTTP) and MgCl2 were purchased from Pharmacia (Alameda, Calif). 2'-Deoxycytosine-5'-O-(1-thiophosphate) (dCTPαS), BsoB1 restriction endonuclease, Bst polymerase and bacterial DNA were supplied by Becton Dickinson (Sparks, Md). Purified human genomic DNA was obtained from Oregon Health Sciences University (Portland, Ore). Oligonucleotides were synthesized by Oligos, Etc. (Wilsonville, Ore). BODIPY Texas Red (BTR) was purchased from Molecular Probes (Eugene, Ore).
Sda Amplification
Two different sets of SDA amplification reactions were developed for the bacterial 16S rRNA gene and for the human Factor V gene. Amplification conditions and concentrations were adapted from previous work10 with the following change: 5'–3' exo-Bst polymerase replaced exo-BCA polymerase. For 16S amplification, 25U/reaction (Bst), 60 U/reaction (BsoB1), and 1 µg bacterial DNA was used. Factor V amplification used 15.6 U/reaction (Bst), 40 U/reaction (BsoB1), and 0.1 µg (Factor V) of genomic DNA obtained from blood. Total reaction volume was 50 µL. Oligonucleotides used for amplification reactions are shown in Table 1. As indicated, Factor V amplicons were constructed using a biotinylated 5' amplifying primer and one of two allele-specific 3' primers. Allele-specific reactions were done separately. Reactions were allowed to proceed for 30 minutes at 60°C and then terminated by the addition of 10 µL of 100 mmol/L ethylenediaminetetraacetic acid (EDTA) and then stored at -20°C. Before electronic targeting, crude amplification reactions were either spun for 2 minutes through G6 columns (Biorad, Hercules, Calif) preequilibrated with distilled water or dialyzed in multiwell plates (Millipore, Bedford, Mass) for at least 5 hours against distilled water. The prepared samples were then mixed in a 1:1 ratio with 100 mmol/L histidine and heated at 95°C for 5 minutes.
SDA amplification primers.
On amplifying primers, BsoB1 recognition sites are boldfaced, genomic homology regions are underlined, and Factor V allele specific 3' termini are shown in boldfaced type.
Bacterial 16S positions refer to a derived consensus sequence; human Factor V sequence refers to GenBank accession #L32764.
Bs indicates sense bumper primer; Ba, antisense bumper primer; As, sense-amplifying primer; Aa, antisense-amplifying primer; Awt, antisense-amplifying primer-wild type; Am, antisense-amplifying primer-mutant.
Factor V Intron 10 (ref. 20).
Oligonucleotides used for bacterial 16S amplicon microarray analysis.
Nucleotides that differ between the bacterial species are boldfaced.
Positions are relative to a derived consensus sequence.
Bio indicates biotin conjugation.
Electronic Microarray Analysis
Electronic targeting of biotinylated oligonucleotides and hybridization of amplicons or reporter oligonucleotides used conditions and microelectronic arrays previously described.8,9 16S: following electronic placement of biotinylated capture oligonucleotides (Table 2), heat-denatured amplicons were targeted to specific array locations. Electronic hybridization of the amplicons was performed, followed by electronic stringency, if appropriate.8 Fluorescently labeled oligonucleotide reporter oligonucleotides were then added in 6X SSC (1X = 0.15 M NaCl, 15 mmol/L sodium citrate, pH 7.0) (Table 2). Passive hybridization was allowed to proceed for 30 minutes at room temperature. The chips were then washed 5–8 times using 0.1X STE/1% sodium dodecyl sulfate followed by 1X STE (1X STE = 10 mmol/L Tris chloride, pH 7.5, 10 mmol/L NaCl, and 1 mmol/L EDTA, pH 8.0). Factor V: 10 µL of each desalted and heat-denatured biotinylated amplification reaction mixture was electronically targeted to specific array microelectrodes.13 A fluorescent labeled reporter oligonucleotide [BTR-dCTGTATTCCTCGCCTGTC] (276–259, GenBank accession #L32764) was introduced in 6XSSC and allowed to hybridize for 30 minutes at room temperature. The array was then washed in 0.1XSTE/1%SDS followed by 1XSTE, and then the resultant fluorescence imaged using laser illumination at 594 nm.
Gel Electrophoresis
Amplification reactions were analyzed using standard protocols14 using either 1% agarose gel or 6% polyacrylamide mini gels (Novex, San Diego, Calif) followed by ethidium bromide staining. Digital images were obtained with a Chemimager gel imaging system (Alpha Innotech, San Leandro, Calif).
Results
Pathogen Screen
In designing assays that enable screening for multiple pathogens, one approach is to focus on highly conserved genes that contain limited regions of divergence between species. One then uses a common amplification platform and sorts for the presence or absence of particular strains or species among the amplicons produced following a common or “universal” amplification reaction. Alternatively, one can select marker genes unique to each species or serotype. Product amplicons themselves then serve as unique indicators for the presence or absence of particular pathogens. Screening objectives and technology determine the suitability of either approach. In order to more fully demonstrate the utility of this screening platform for multiple gene products, we used the former approach and targeted our screening analysis to the highly conserved bacterial gene encoding for 16S rRNA.
Despite high overall sequence conservation, there are regions of microheterogeneity within 16S that are extremely useful for discrimination between closely related bacterial species.15 The bracketing of these microheterogeneities by conserved sequences provides the opportunity to design primers for consensus amplification encompassing almost all bacterial species. The region chosen for this study corresponds to the highly conserved loop III portion of 16S rRNA. Contained within this region is a stretch (> 20 nucleotides) of highly polymorphic sequence flanked by more conserved regions. This divergent region was used to discriminate between the bacteria examined. Related studies using PCR as a means of target amplification have been reported, demonstrating the general utility of the 16S gene for this purpose.16,17 In the present study, we chose oligonucleotides to distinguish between four pathogenic bacteria isolates selected from ATCC. Others could be chosen to encompass the range of pathogenic and nonpathogenic bacteria potentially encountered in a clinical setting.
Purified DNA from four pathogenic bacteria (E. coli O157:H7, Salmonella typhimurium, Shigella dysenteriae, or Campylobacter jejuni) were amplified by the isothermal amplification technique, SDA, 10 using a common set of 16S “consensus” primers. Product amplicons were then resolved using agarose gel electrophoresis to evaluate amplification efficiencies. Representative results comparing three amplification reactions, E. coli, Shigella typhimurium, and Shigella dysenteriae are shown (Figure 1) and indicate similar levels of amplification efficiency for each. DNA from Campylobacter jejuni also yielded similar levels of amplification (data not shown), thereby providing amplicons from all four bacteria with which to conduct the subsequent analysis.
Multiple forms of assay are possible using the electronic microarray platform. Two basic forms were used in this portion of the study, both based on a sandwich assay format. 18 The first of these uses localization of a common binding or capture oligonucleotide over several array sites followed by electronic hybridization of the bacterial amplicon. Identification of bacterial type is then made using type or sequence-specific fluorescently labeled reporter oligonucleotides (Table 2).
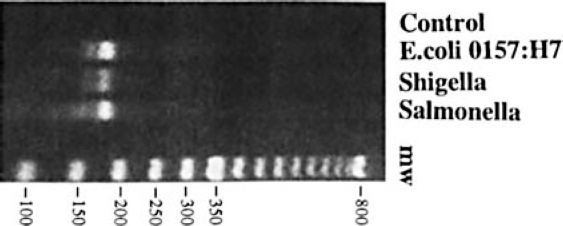
16S SDA amplicons purified DNA from E. coli, Shigella, and Salmonella. DNA was amplified by SDA using the same set of amplifying primers, as described in Methods. A portion of the crude reaction was then subjected to agarose gel electrophoresis and product amplicons visualized using ethidium bromide. Control = reaction minus template.
In this first approach, universal capture oligonucleotides were electronically targeted to specific locations upon the microelectrode array. Electronic hybridization between sample amplicon and capture oligonucleotide was then done at each site. Several advantages arise from the use of electronic targeting and hybridization.8,9 The first is that oligonucleotides and amplicons can be targeted to select locations on the microelectronic array, allowing diverse materials to be positioned selectively. A second advantage is that hybridization is accelerated by concentration at the selected microelectrode, ie, by increased mass action. Third, because of the absence of free cations (required for phosphate backbone shielding) in buffers used for electronic hybridization, denatured amplicons remain single-stranded until reaching the appropriate location and electronic hybridization environment. Thus, in the bulk solution, there is effectively no rehybridization of denatured double stranded materials and thereby minimal loss of signal due to removal of single stranded target from bulk solution. Finally, electronic control also allows electronic stringency to be used for the removal of poorly matched hybrids, if so desired.
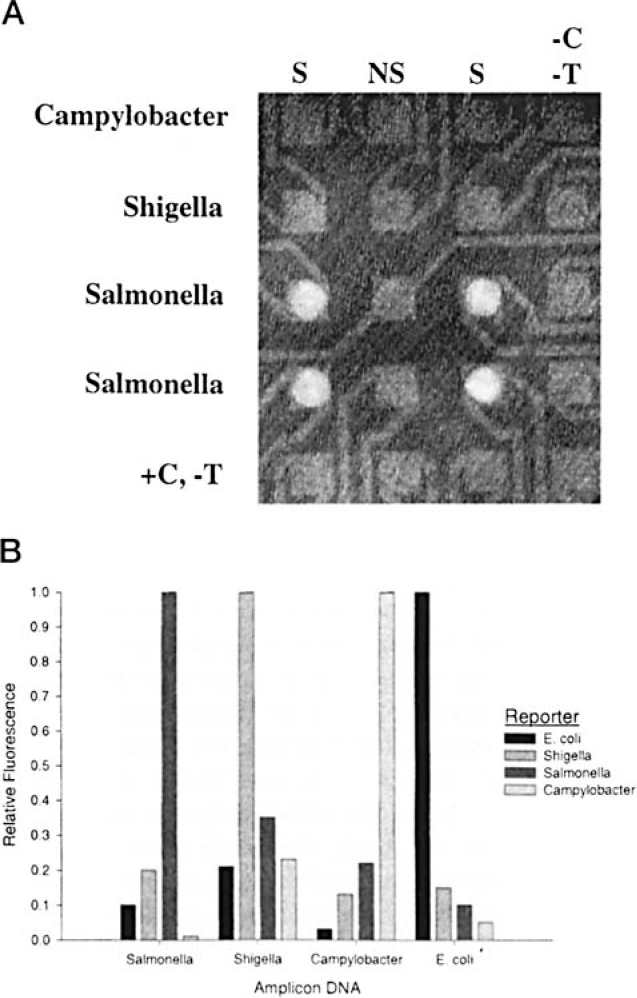
16S assay using common capture and specific reporter oligonucleotides. A, Example hybridization using multiple bacterial amplicons and Salmonella-specific reporter. As described in Methods, amplicons from Campylobacter, Shigella, or Salmonella were electronically hybridized to the indicated rows. Salmonella-specific fluorescently labeled reporter was then hybridized to all sites on the microarray. Following washing, the resultant fluorescence was imaged as shown and quantified. S indicates universal capture; NS, nonspecific capture oligonucleotide; -C,-T, no capture, no target addressed; +C,-T, capture addressed but no target addressed. B, Discrimination of enteric bacteria using specific reporters. Shown are the compiled results from four experiments where SDA amplicons from four bacterial types were electronically addressed to individual sites on microarrays and sequence-specific BTR-labeled reporter probes were passively hybridized, in a fashion similar to that shown in A. Results from each set of experiments were normalized to the maximal signal observed within each test group.
The results of a typical assay are shown in Figure 2a. In this experiment, the universal 16S capture oligonucleotide was electronically targeted to the array columns indicated (S). A different, unrelated oligonucleotide was targeted to the microelectrodes in between these two columns (NS). Each type of bacterial amplicon was sequentially targeted and hybridized electronically at each indicated row of microelectrodes, taking advantage of the selective placement gained when using electronically controlled microarrays mentioned above. A fluorescently labeled reporter oligonucleotide specific for Salmonella was then passively hybridized at all sites. This was followed by a low salt wash and a fluorescent image of the array taken. As shown, the strongest signal is obtained from those sites where both specific 16S capture oligonucleotides and Salmonella amplicons were targeted. The absence of either of these resulted in either a weak or an undetectable signal.
As shown in Figure 2b, high specific/nonspecific discrimination ratios were obtained using this assay format for each of the bacteria (between 3-10 depending on the target and the reporter combination). Given that a common fluorescent dye was used for the detection schemes, serial application of reporter oligonucleotides needed to be used to evaluate the presence or absence of all four bacteria. Other variations on this assay format, eg, multiple wavelength dyes with appropriate lasers, presumably would make serial application of different reporters unnecessary.
The second assay format is the converse of that described above. That is, discrimination between bacterial types is done by use of sequence-specific capture oligonucleotides prepositioned on the array. Target amplicons are electronically addressed to each site on the microarray. This hybridization step is immediately followed by electronic stringency to remove mismatch hybrids. Following application of a common or universal fluorescently labeled reporter oligonucleotide, identification of bacterial type is then made by array location. So, in addition to the selectivity of oligonucleotide and target placement on the microarray mentioned above, the additional advantage of electronic stringency to remove mismatched hybrids is used in this assay format.
Results from a demonstration of this assay format are shown in Figure 3. In this experiment, 16S amplicons from Salmonella were electronically targeted to type-specific capture oligonucleotides (Table 2). Electronic stringency 13 was applied to all pads and then the universal reporter oligonucleotide was added and the resultant fluorescence at each site on the array quantified. In general, this approach provides for higher discrimination ratios between specific and nonspecific signals than the alternate methodology (Figure 2b). This suggests that electronic stringency provides for more facile and controlled discrimination between matched and mismatched hybrids than does traditional methods, and is consistent with our earlier findings.8,13
Genetic Predisposition Screen
The genetic screening examines for the presence or absence of the human Factor V Leiden mutation. The Leiden mutation is a single point mutation found near the terminus of exon 10 of the Factor V gene whose homozygous or heterozygous presence leads to a predisposition to deep venous thrombosis.19,20 In order to provide additional point mutation assays, in addition to those electronic microarray assays previously demonstrated,8,13 allele-specific strand displacement amplification was developed. To selectively amplify either the normal or mutant genotype, the SDA amplifying primers in the antisense orientation were designed with their 3' terminus complementary to either the wild type nucleotide base, G, or the point mutation base, A, present in exon 10 of the factor V gene (Table 1). The corresponding sense primer was common in both reactions. However, this primer was modified by incorporating a biotin moiety on its 5' end in order to provide a facile mechanism for capturing the amplicon on the array following electronic targeting. 13 Following amplification and position-specific targeting of each amplification reaction, the array was evaluated in a site-specific fashion for the presence or absence of targeted amplicons. Individual electronic control of each test pad allowed analysis of multiple samples upon the microarray.
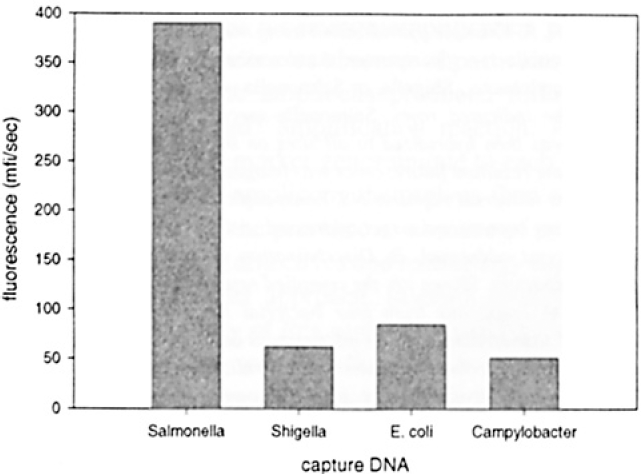
Example of 16S assay using specific captures and a common reporter oligonucleotide. As described in Methods, Salmonella DNA was amplified then electronically targeted to an array containing sequence-specific capture oligonucleotides. Electronic stringency was then applied to remove mismatched hybrids and then a common fluorescently labeled reporter oligonucleotide was used to visualize signal upon the array. Quantitated fluorescence present at each array location is shown. mfi/sec indicates mean fluorescence intensity per second.
Eighteen clinical blood DNA samples were analyzed without prior knowledge of the patient's Leiden mutation status. Two allele-specific SDA reactions per sample (containing either normal or mutant primers) were conducted in parallel to examine each patient's genotype. The PAGE results from five of these pair-wise reactions are shown in Figure 4. As can be seen, the allele-specific amplification reactions under these conditions is highly specific. That is, the selective absence of visible mutant or normal-type amplicons suggests that the amplification reaction is sensitive to the presence or absence of the Factor V Leiden mutation in these individuals.
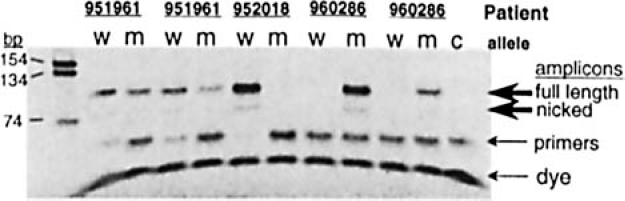
Factor V Leiden mutation detection in multiple patients. Genomic DNA samples were amplified with allele-specific SDA amplification using two separate reactions each (for either the normal genotype or for the Leiden mutation), as described in Methods. Shown are the polyacrylamide gel analyses of the allele-specific reactions using DNA from three patients (two of which are duplicate assays). w indicates normal-genotype amplification; m, mutant genotype amplification; and c, no template control.
All amplicon reactions, regardless of the presence or absence of amplified material by gel analysis, were uniformly treated and sequentially targeted to specific locations upon the array. Representative results from three DNA patient samples are shown in Figure 5. Each of these samples were targeted in duplicate. The presence or absence of a fluorescent signal from a hybridized complementary reporter oligonucleotide to a conserved sequence indicated the presence or absence of Factor V amplicons. As shown, hybridized signals were several-fold greater than the nonaddressed background signal. Also, signal from putatively positive mutant samples was lower then that from wild type amplicons (Figure 5). Overall, microelectronic assay results correlate 100% with the gel results shown in Figure 4.
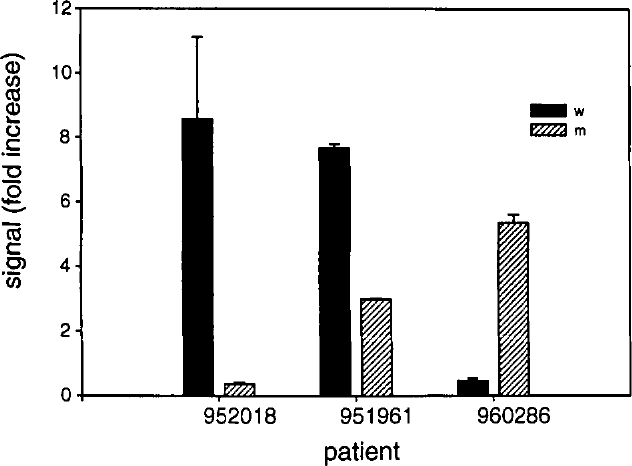
Representative histogram from electronic array analysis of Factor V SDA amplicons. As described in Methods, DNA from three patients were amplified using either mutant or wild type-specific primers. Following electronic addressing to specific array locations, a reporter oligonucleotide was added and fluorescence at each location quantified. Results shown are the average of the duplicate addressing (error bars indicate range). Fold increase indicates level of fluorescence signal as compared to that observed at non-addressed array sites. Abbreviations as in Figure 4.
It was interesting to note that the strength of the fluorescent signal approximated the apparent quantity of amplified material. This was most striking in those samples with a less efficient amplification reaction, such as was seen with the DNA from patient 951961 in Figure 4. To accommodate apparent amplification differences, scoring of sites was simply made by making the criteria for a positive signal to be at least 2-fold above background signal present at nonaddressed sites. As shown in Table 3, there was 100% correlation between the presence of amplified material by gel analysis and the presence of positively scored signals upon the array. In addition, duplicate assays performed on the same patient sample yielded consistent findings. In short, these results support the concept that multiple sample analysis by the serial application of samples followed by single reporter detection is possible using a microelectronic array and may serve to supplement or replace other forms of analysis, eg, gel electrophoresis, in applications such as this.
Because these samples were analyzed before knowledge of their mutational status, it was of interest to determine if the apparent allele specificity of the amplification reaction did, in fact, correspond with clinical status. In all, 18 individuals were examined: 6 homozygous wild type, 6 heterozygous, and 6 homozygous mutant. One each from these three groups was analyzed in duplicate. As shown in Table 3, the selectivity of the allele-specific amplification reaction was in complete agreement with the mutational status of each sample as determined by PCR-RFLP analysis. 19 Thus, combined with allele-specific amplification using SDA, analysis of amplicon product formation upon an electronic array may be a useful method for analysis of the Factor V Leiden mutation from multiple patient samples.
Discussion
We have demonstrated two model clinical assays combining amplified materials and microelectronic arrays, one potentially suitable for pathogen screening and the other for a deleterious genetic mutation. To date, tests such as these are not routinely used. One reason is that until recently, oligonucleotide-based DNA detection methodologies have been restricted by the frequently long amplification and hybridization times required to achieve resolvable signals. An additional limitation to these methodologies is the inability to multiplex different hybridization events upon a single analytical surface, thereby restricting information obtainable in any one assay. Both of these limitations are overcome by the union of SDA with active microelectronic arrays capable of selectively targeting and concentrating DNA to specific array locations. A further strength of these devices is the power to perform electronic denaturation allowing discrimination of single base polymorphisms.8,13 Thus, studies such as these move biodiagnostics closer to the goal of rapid, low-cost, and patient-tailored DNA-based diagnostics.
In general, today's generation of microarray technologies require an amplification step either of the target DNA itself or of the resultant signal. Target amplification offers two distinct advantages. First, it provides increased specific material thereby lessening signal sensitivity needs and, second, it effectively reduces the level of extraneous or potentially interfering DNA. Therefore, the ability to combine an efficient amplification methodology with rapid and specific detection provides a powerful synergy in clinical microassays. Toward this end, we have examined a nonpolymerase chain reaction (non-PCR) amplification technique, SDA. SDA is an amplification methodology that has the sensitivity and robustness to rapidly (eg, 15 to 30 minutes) and exponentially amplify a small number of target molecules from a complex background. 10 However, in contrast to PCR, SDA is an isothermal technique that affords simpler thermal control and associated instrumentation. As such, it is more compatible with a unified amplification-hybridization/detection system for rapid analyses of nucleic acids.
Allele-specific factor V SDA amplification results.
X indicates positive; O, negative.
Genotype was determined by PCR-RFLP using Mnl-1 restriction endonuclease. 19
To extend the capabilities of this amplification technology, we report for the first time allele-specific amplification using SDA. In general, the results appear equivalent to allele- or sequence-specific PCR for Factor V mutation analyses. 21 This is understandable because key to both is the extension by a polymerase from the hybridized amplification primer. The specificity of this process comes from the inability to extend efficiently when the 3' end nucleotide of the primer is noncomplementary to the target sequence. The robustness of Factor V allele-specific PCR can be increased by primer modification or by use of peptide nucleic acid clamping.22,23 It remains to be seen whether allele-specific SDA can be likewise improved. Such levels of sensitivity would be suitable for simple allelic analysis as described here. In addition, the use of a control amplification reaction run within these reactions using a different wavelength fluorescent reporter would guard against false negatives due to failed components or the presence of inhibitors. However, it may be that for other applications, eg, such as analysis of mutations developed during oncogenesis, high background levels of nonmutated DNA may preclude allele-specific amplification. It is clear that SDA, allele-specific or otherwise, combined with active microchip technology offers a powerful combination of technologies and may offer a glimpse of the next phase of biodiagnostics.
Footnotes
Acknowledgments
We would like to thank Ronald Sosnowski, Michael Heller, James O'Connell, Tina Nova, and Dan McLauren for their encouragement and helpful suggestions.