
Abstract
Background
Very small preterm infants who have genital mycoplasmas isolated from the trachea are at increased risk to develop bronchopulmonary dysplasia (BPD). The early stages of BPD are characterized by inflammation. Recruitment and activation of mononuclear cells in response to mycoplasmas may be important in the early stage of the disease. Lung epithelial cell production of monocyte chemoattractant protein-1 (MCP-1), a protein that attracts and activates mononuclear cells, could be critical in the regulation of mononuclear cell migration to the lung.
Methods
We examined the potential of Mycoplasma hominis (Mh) to induce MCP-1 gene expression and protein production in A549 cells, a pulmonary epithelial cell line with characteristics of type II cells.
Results
Live or heat-inactivated Mh induces MCP-1 mRNA and protein in a dose- and time-dependent manner. Stimulation of MCP-1 by Mh was not inhibited by 50 µg/mL of polymyxin B, interleukin (IL)-1ra, or neutralizing antibodies to IL-1a, IL-1β, or tumor necrosis factor a (TNF-α). IL-1a, IL-1β, and TNF-a were not detected in conditioned media of Mh-stimulated A549 cells.
Conclusions
These data suggest that Mh may participate in the inflammatory component of BPD by directly inducing epithelial cellproduction of cytokines that recruit and activate mononuclear cells.
Introduction
Inflammation indicates the early stages of bronchopulmonary dysplasia (BPD) and is characterized by the recruitment of polymorphonuclear leukocytes and mononuclear cells to the lung.1–3 Recruitment of inflammatory cells to a site of injury is controlled by locally produced chemoattractant chemokines such as interleukin-8 (IL-8), epithelial-derived neutrophil-activating peptide-78 (ENA-78), and monocyte chemoattractant protein-1 (MCP-1). 4 The role of chemokines in inflammation and lung injury that results in BPD is suggested by increased concentrations of IL-8, IL-1β, tumor necrosis factor-α (TNF-α), and macrophage inflammatory protein-1a in the tracheal aspirates of infants who subsequently developed BPD.2,5–7 Although polymorphonuclear leukocytes are the predominate cells in the tracheal aspirates of these infants early in the disease, recruitment of mononuclear cells to the lungs and their subsequent activation is a significant component of the inflammatory response.
Alveolar type II cells constitute approximately 15% of the cells in the lung. 8 Situated between the alveolar space and the interstitium, type II cells are ideally located to mediate the traffic of immune cells into the alveoli. Recent studies demonstrate that respiratory epithelial cells participate in inflammatory responses of the lung by the elaboration of the chemotactic cytokines IL-8, ENA-78, and MCP-1 in response to both macrophage-derived cytokines (eg, IL-1 and TNF-α) and respiratory pathogens.9–19
Although the factors that initiate the inflammatory component in BPD are not understood, pulmonary infections are potentially important cofactors in the development of this disease. Genital mycoplasmas frequently are found in the airways of small preterm infants and are associated with a high risk of subsequently developing BPD.20,21 We have demonstrated previously that Mycoplasma hominis (Mh) can induce the production of neutrophil chemoattractants, such as IL-8 and ENA-78, by respiratory epithelial cells. 14 Moreover, we have shown that the concentration of MCP-1 is significantly increased in the tracheal aspirates of infants who develop BPD and is associated with the isolation of Mh and Ureaplasma urealyticum (Uu) from tracheal aspirates. 22 Because mononuclear cell recruitment and activation are important components of BPD, we designed experiments to determine whether respiratory epithelial cells produce MCP-1 in response to Mh.
Materials and Methods
Cytokines and Antibodies
Recombinant human MCP-1, monoclonal mouse (immunoglobulin G1 [IgG1]) antihuman MCP-1 antibodies, and polyclonal goat (IgG) antihuman MCP-1 antibodies were purchased from R&D Systems (Minneapolis, Minn). For neutralizing polyclonal antibodies to human IL-1α, IL-1β, and TNF-α, monoclonal and biotinylated monoclonal antihuman IL-1β and TNF-α antibodies (for use in enzyme-linked immunoadsorbent assay [ELISA]) were obtained from Endogen (Cambridge, Mass). Recombinant human IL-1β, IL-1α, TNF-α, and IL-1 receptor antagonist (IL-lra) were also obtained from Endogen. Peroxidase-conjugated polyclonal rabbit (IgG) antigoat IgG detecting antibody was obtained from Bio-Rad laboratories (Hercules, Calif). Polymyxin B was obtained from Sigma (St Louis, Mo).
Cell Culture
A549 lung epithelial cells were obtained from American Type Culture Collection (ATCC, Rockville, Md). Cells were maintained in F12K media containing 10% fetal calf serum (FCS) at 37°C in 5% CO2.
Bacterial Culture
Mh, serotype 5, (ATCC #23114) was grown in arginine-enriched pleuropneumonia-like organism broth (AB) for 48 hours at 37°C in 5% CO2. 23 Mh was harvested and concentrated by centrifugation for 15 minutes at 14,000g. The Mh pellet was reconstituted in fresh AB, and aliquots were prepared for cryopreservation in liquid nitrogen by addition of 20% glycerol. The Mh stock used for viable Mh studies contained approximately 7.6×107 Mh/mL after recovery from liquid nitrogen as determined by titration in AB. A heat-inactivated Mh stock, equivalent in titer to the viable Mh stock, was prepared by exposing viable Mh (108 Mh/mL) to 95°C for 10 minutes. This stock was used as a heat-inactivated Mh antigen preparation. Lethality was confirmed by the failure of Mh to grow after 7 days of culture in AB broth. Conditioned AB was prepared by filtering a 72-hour culture of Mh through a 0.1-µm filter. No growth was observed in this filtrate after 7 days in culture.
Experimental Conditions
Cells were plated in 24 well plates at 2×105 cells/ well/mL in F12K (10% FCS) and grown to confluence at 37°C in 5% CO2. The media from confluent monolayers was removed, and quadruplicate wells were either a) infected with 3.8×106 Mh/well (100 µL; multiplicity of infection, ~10:1) for 1 hour at 37°C or b) stimulated for 48 hours with a heat-killed Mh preparation equivalent to 108 bacteria/mL. After allowing viable Mh to attach to the cell monolayers, fresh antibiotic-free F12K (1% FCS) was added to the wells to a final volume of 1 mL and incubated at 37°C in 5% CO2 for 48 hours. Recombinant human IL-1β (50 pg/mL) was added to quadruplicate A549 culture wells (without mycoplasma) as a positive control inducer of MCP-1. In some experiments, 100 µL of conditioned AB broth was examined for its potential to stimulate cytokine production. Culture media were collected and frozen until MCP-1 concentrations could be determined by ELISA. To assess whether lipopolysaccharide (LPS) contamination of the Mh preparations might contribute to MCP-1 induction, polymyxin B (50 µg/mL) was co-incubated with heat killed Mh (~5×106 organisms/mL). After 48 hours, the conditioned media was harvested and frozen at −70°C for subsequent determination of MCP-1 by ELISA.
To rule out the possibility that autocrine production of IL-1α, IL-1β, or TNF-α, as opposed to Mh, is responsible for the induction of MCP-1, A549 cells were incubated with IL-1ra (100 ng/mL) or neutralizing antibodies to IL-1α (11 µg/mL), IL-1β (10 µg/mL), and TNF-α (11.1 µg/mL) before the addition of heat-killed Mh (~5×106 organisms/mL). We had previously determined that these concentrations of blocking antibodies or IL-1ra inhibited the production of MCP-1 in A549 cells by greater than 95% with 50 pg/mL of IL-1β, 100 pg/mL of IL-1α, or 10 ng/mL of TNF-α (data not shown) After 48 hours, the conditioned media was harvested and frozen at −70°C for subsequent determination of MCP-1 by ELISA.
The effect of the Mh on MCP-1 mRNA expression was assessed by qualitative reverse transcriptase polymerase chain reaction (RT-PCR). Total RNA was isolated from pooled quadruplicate wells treated as described above and steady-state (48-hour) MCP-1 mRNA assessed by RT-PCR. To assess kinetics of MCP-1 mRNA, A549 cells were grown to confluence in six-well tissue culture dishes. One day before the experiments, the media was changed to F12K with 1% FCS. Fifty microliters of heat-killed Mh (equivalent to 5×10 6 organisms) were added to each well and incubated for 15, 45, 60, or 120 minutes. Total RNA was isolated from untreated cells and from cells treated with heat-killed Mh for the described durations, and MCP-1 mRNA was assessed by RT-PCR.
Elisa
MCP-1 concentrations were determined by ELISA. Antibody concentrations were titrated for optimal performance. ELISA plates were prepared as follows. Plates were coated overnight at 4°C with 100 µL of monoclonal antibody to MCP-1 (1.3 µg/mL) in 0.1 mM sodium carbonate buffer (pH 9.6). After coating, ELISA plates were washed three times with 200 µL of phosphate-buffered saline (PBS) containing 0.05% Tween-20 (wash buffer) then blocked using 200 µL PBS-Tween-20 containing 3% (wt/vol) bovine serum albumin (blocking buffer). After blocking, the plates were washed three times before use. Assays were performed in blocking buffer. Culture media and recombinant cytokine standards were diluted as appropriate in blocking buffer. For the MCP-1 ELISA, 100 µL/well of culture supernatant or recombinant cytokine standards were incubated for 2 hours at room temperature. The plates were then washed three times as previously described and incubated overnight at 4°C with 100 µL/well of the appropriate polyclonal antihuman MCP-1 antibodies at 1.3 µg/mL in blocking buffer. Thereafter, the plates were washed three times and incubated for 2 hours at ambient temperature with 100 µL/well of blocking buffer containing polyclonal rabbit antigoat IgG peroxidase-conjugate at a 1:1000 dilution. The plates were then washed three times. The enzyme substrate 2,2′-azino-di[3-ethyl-benzthiazoline-6-sulphonic acid] was used to quantify the amount of MCP-1. The reaction was stopped with 100 µL of 2% oxalic acid, and plates were read at 405 nm. Sensitivity of this assay was 300 pg/mL.
IL-1β concentrations were determined by ELISA. Plates were coated overnight at 4°C with 100 µL of monoclonal antibody to IL-1β (3.4 µg/mL) in 0.1 mM sodium carbonate buffer (pH 9.6). After coating, ELISA plates were washed and blocked as described above. After blocking, the plates were washed three times before use. Fifty microliters/well of culture supernatant or recombinant cytokine standards was incubated for 1 hour at room temperature. Fifty microliters/well of biotinylated monoclonal antihuman IL-1β antibody (208 ng/mL in blocking buffer) was then added and the plate incubated for an additional hour at room temperature. After washing the plate four times, 100 µL/well of a poly-horseradish-peroxidase streptavidin conjugate (Pharmingen, Inc, San Diego, Calif) was added and the plate incubated for 30 minutes at room temperature. After washing, the substrate was added and the concentrations colorimetrically determined as described above. The sensitivity of this assay was 7.8 pg/mL.
TNF-α concentrations were determined by ELISA. Plates were coated overnight at 4°C with 100 µL of monoclonal antibody to TNF-α (3.5 µg/mL) in PBS (pH 7.4). After being coated, ELISA plates were washed and blocked as described above. After being blocked, the plates were washed three times before use. Fifty microliters/well of culture supernatant or recombinant cytokine standards was incubated for 1 hour at room temperature. Fifty microliters/well of biotinylated monoclonal antihuman TNF-α antibody (500 ng/mL in blocking buffer) was then added and the plate incubated for an additional hour at room temperature. After the plate was washed four times, 100 µL/well of the poly-horseradish-peroxidase streptavidin conjugate was added and the plate incubated for 30 minutes at room temperature. Thereafter, substrate was added and the concentrations colorimetrically determined as described above. The sensitivity of this assay was 7.8 pg/mL.
IL-1α concentrations were determined by use of a commercial ELISA kit (R&D Systems). The assay was performed according to the manufacturer's specifications. The sensitivity of this assay was 3.9 pg/mL.
Total RNA Isolation
Isolation of total RNA was performed with the RNeasy Mini kits from Qiagen (Chatsworth, Calif) according to the manufacturer's specifications. After the culture media for ELISA analysis was removed, the remaining quadruplicate cell monolayers were lysed and applied to silica gel spin columns, which were washed in appropriate buffers and total RNA eluted using 50 µL of Rnase-free water. The total RNA recovered from each sample was quantified and assessed for purity by spectrophotometric analysis at 260 nm and 280 nm. Equivalent amounts (1 µg) of total RNA were then subjected to RT-PCR analysis to assess the relative concentrations of cytokine-specific mRNA between treatments.
Primers
The oligonucleotide primers used for RT-PCR were synthesized by Integrated DNA Technologies, Inc (Coralville, Iowa). The PCR primer set for MCP-1 consisted of a 22-base sense primer (5′-TTCTGTGCCTGCTGCTCATAGC-3′) and a 22-base antisense primer (5′-TCCTGAACCCACTTCTGCTTGG-3′). 10 The PCR primer set for β-actin was a 21-base sense primer (5′-CTG GCA CCC AGC ACA ATG AAG-3′) and 20 base antisense primer (5′-ACC GAC TGC TGT CAC CTT CA-3′). 24 Reverse transcription of total mRNA into cDNA was performed using a 20-base oligo-dT primer.
Pcr
The mRNA from 1 µg of total RNA was reverse transcribed into single-stranded cDNA and MCP-1-specific cDNA amplified by PCR using an Access RT-PCR System kit (Promega, Madison, Wis). The concentration of each primer was approximately 0.16 µM in a 50-µL reaction mix. Cycling parameters consisted of an initial oligo-dT (1 μΜ) primed reverse transcription step for 1 hour at 37°C. The avian myeloblastosis virus reverse transcriptase was then inactivated for 2 minutes at 94°C. The CDNA transcripts were then amplified for 40 cycles, each cycle consisting of an initial melting step at 94°C for 30 seconds, annealing at 58°C for 1 minute, and extension at 68°C for 2 minutes. All PCR amplifications included a final extension step at 68°C for 10 minutes to ensure full length transcripts. Initially, samples of the PCR products were taken at 15, 20, 25, and 35 cycles to determine the minimal number of cycles required to detect products and to optimize differences between treatment groups. As a control, we examined the β-actin mRNA in A549 cells to demonstrate a specific effect on MCP-1 gene transcription by Mh or Mh antigen. RT-PCR conditions for β-actin were identical to those used for MCP-1, except that 0.5 µg of total RNA was used and the primer concentrations were 0.5 µM. The amplicons were visualized by agarose gel electrophoresis.
Agarose Gel Electrophoresis
The RT-PCR products were visualized on ethidium bromide stained 2% agarose gels. Briefly, 10 µL of each RT-PCR reaction was diluted 1:1 in 10 µL sample buffer (6X) containing 40% wt/vol sucrose and 0.25% wt/vol bromophenol blue as a tracking dye and was loaded on 2% agarose gels before electrophoresis. Electrophoresis was performed in Tris-acetate-EDTA buffer at 100 V for 1.5 hours. Gels were stained with 0.5 µg/mL ethidium bromide, and the cytokine-specific amplicons were visualized using a ultraviolet transilluminator. The molecular weight of the RT-PCR products was confirmed by comparison with a DNA ladder that consisted of DNA fragments ranging from 100 to 1000 bp in 100 bp increments (Promega).
Statistical Analysis
All statistical analyses were performed using the SPSS for Windows version (SPSS, Inc, Chicago, III). Statistically significant differences in cytokine concentration between the experimental and control cultures were determined by one-way analysis of variance followed by the Student-Newman-Keuls test of multiple means. P values less than 0.05 were considered significant. Data are reported as the mean±SEM.
Results
Figure 1 illustrates the effect of Mh on the production of MCP-1 by A549 type II epithelial cells as determined by ELISA. Mh infection of type II cells with 3.8×106 Mh/mL/well (multiplicity of infection, 10:1) resulted in a significant increase in production of MCP-1 (Figure 1), when compared with either untreated controls (Control) or cultures incubated with AB. Infection of cultures with Mh for 48 hours caused MCP-1 concentrations to increase from 2.4±0.6 ng/mL (Control) to 11.1±2.0 ng/mL (Mh). Additionally, there was no significant difference in MCP-1 concentrations between untreated controls and those cultures that received AB, a cell-free media used for culturing Mh. IL-1β (a known inducer of MCP-1) significantly increased MCP-1 production. Coincubation with 50 µg/mL of polymyxin B did not affect induction of MCP-1 by heat-killed Mh (control, 1.4±0.1 ng/mL; heat-killed Mh, 4.1±0.2 ng/mL; Heat-killed Mh plus polymyxin B, 4.9±0.4 ng/mL; P=not significant).
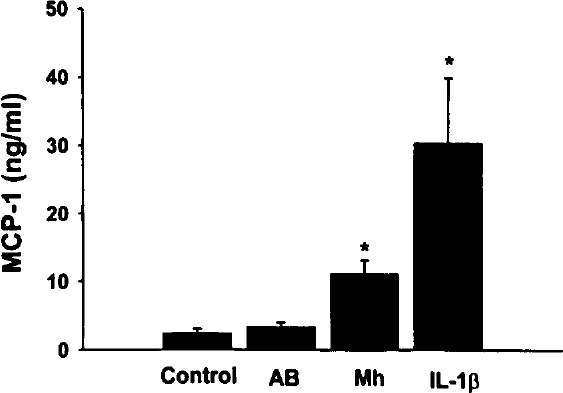
Mh induction of MCP-1 in A549 cells. Quadruplicate wells of confluent A549 cells were untreated (Control) or cultured with either fresh AB, Mh at 3.8×106 Mh/mL, or 50 pg/mL IL-1β for 48 hours at 37°C, 5% CO2. Culture media were harvested and the MCP-1 levels determined by ELISA. Values represent the mean concentration in nanograms per milliliter ±SEM of 6 experiments. The (*) indicates statistically significant differences when compared with untreated controls.
The kinetics of MCP-1 production after Mh infection of A549 cultures is illustrated in Figure 2. Culture media harvested at 24 hours after infection revealed little change in MCP-1 concentration when compared with controls (Figure 2). However, the concentrations of Mh-induced MCP-1 were significantly greater at 48 and 72 hours after infection than concentrations obtained from untreated 72-hour controls (Control) (Figure 2). The differences observed between Mh-infected cultures harvested at 48 and 72 hours were not statistically significant.
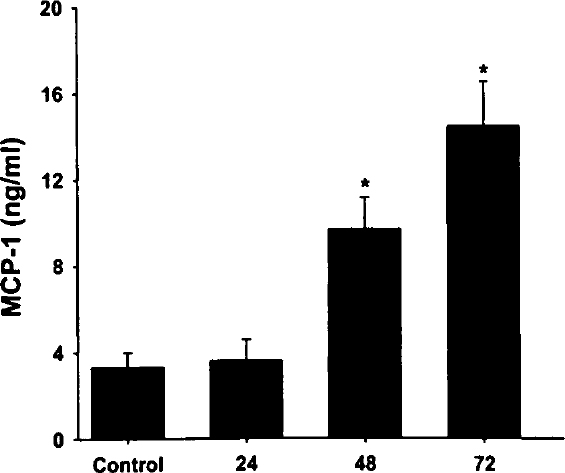
Kinetics of MCP-1 induction by Mh in A549 cells. Quadruplicate wells of confluent A549 cells were untreated for 72 hours or cultured with Mh at 3.8×106 Mh/mL as described. Culture media were harvested at 24, 48, and 72 hours after infection, and the MCP-1 concentrations were determined by ELISA. Values represent the mean concentration in nanograms per milliliter ±SEM of 3 experiments.
Because viable Mh stimulated MCP-1 production in type II cells, we examined the ability of heat-inactivated Mh (~108 Mh/mL) to induce cytokine production. A typical heat-killed Mh dose response curve, shown in Figure 3, illustrates the cytokine production in response to increasing concentrations of heat-inactivated Mh. As can be seen in this figure, the heat-killed preparation caused a dose-dependent increase in MCP-1 production by A549 cells. The MCP-1 concentrations increased from 3.9±0.6 ng/mL in response to 2×105 heat-killed organisms/mL up to 11.1±1.1 ng/mL in cultures that received 5×106 heat-killed organisms/mL (Figure 3). The concentration of MCP-1 produced in cultures stimulated with 5×106 heat-killed organisms/mL represented an approximate 3.5-fold increase above control. The results of these studies suggest that both viable and nonviable Mh (Mh antigen) are capable of stimulating the production of MCP-1 in type II cells. In contrast, conditioned AB broth did not induce MCP-1 production (data not shown)
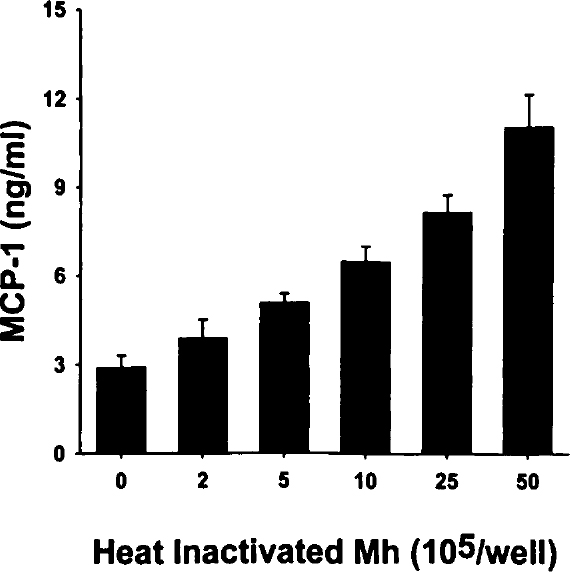
Dose-dependent induction of MCP-1 by heat-inactivated Mh. Confluent A549 cells were stimulated with increasing concentrations (expressed as number of organisms ×105) heat-inactivated Mh for 48 hours. Culture media was harvested and the MCP-1 concentrations determined by ELISA. Values represent a single typical dose response curve and are expressed as the mean concentration ±SD.
We wanted to rule out the possibility that autocrine production of IL-1a, IL-1β, or TNF-α was responsible for the observed induction of MCP-1. Conditioned media from 48-hour incubations with heat-killed Mh did not have detectable levels of IL-1α, IL-1β, or TNF-α. Further, prior incubation of A549 cells with neutralizing antibodies against IL-1α, IL-1β, and TNF-α or IL-1ra did not decrease MCP-1 production induced by heat-killed Mh (Figure 4).
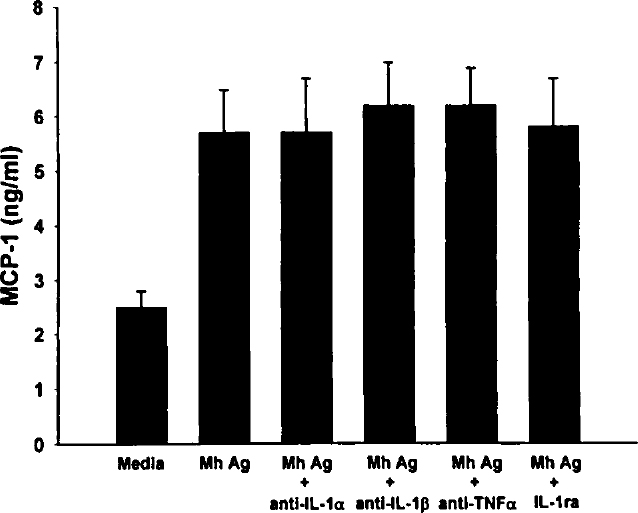
Incubation of A549 cells with IL-1ra and neutralizing antibodies. A549 cells were incubated with neutralizing antibodies to IL-1α, IL-1β, and TNF-α or with 100 ng/mL of IL-1ra before addition of heat-killed Mh (5×106 organisms). Culture media were harvested after 48 hours, and the MCP-1 concentrations were determined by ELISA. Values represent the mean concentration in nanograms per milliliter ±SEM of 4 experiments. Abbreviation: Mh Ag, Mh antigen.
Figure 5 shows the MCP-1-specific mRNA-dependent RT-PCR products from control and Mh-infected A549 cells after 48 hours of incubation. Both viable Mh and heat-killed Mh increased MCP-1 mRNA at 48 hours when compared with controls. Although the MCP-1-specific RT-PCR product (top row) from untreated cultures (Control) was barely visible, infection with viable Mh or stimulation with heat-inactivated Mh resulted in an increase in the amount of MCP-1 RT-PCR product. These results parallel those of the ELISA analysis, which demonstrated low-level basal MCP-1 production in A549 cultures and significantly enhanced production of MCP-1 in cultures that received live or heat-inactivated Mh (Figure 1).
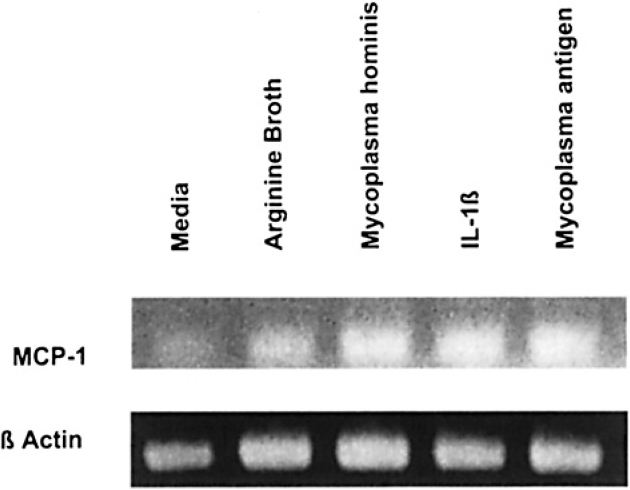
RT-PCR analysis of Mh-induced cytokine mRNA. Shown are the qualitative differences in steady-state MCP-1-specific mRNA-dependent RT-PCR products after electrophoresis on 2% agarose gels and subsequent staining with ethidium bromide. Total RNA was isolated from untreated (control), AB-treated, Mh-infected, IL-1β stimulated or heat-killed Mh stimulated (Mh antigen) A549 cultures, and equal amounts of total RNA subjected to oligo-dT-primed RT-PCR using amplification primers specific for MCP-1 (top), or β-actin (bottom). All RT-PCR products were compared with a standard DNA ladder.
The kinetics of MCP-1 mRNA expression by heat-killed Mh is shown in Figure 6. The MCP-1-specific RT-PCR product (top row) was not detectable in untreated cells or in those treated with heat-inactivated Mh 15 minutes after stimulation. Increased MCP-1 mRNA level was detected after 30 minutes of stimulation, with maximal amounts being detected after 2 hours of stimulation.
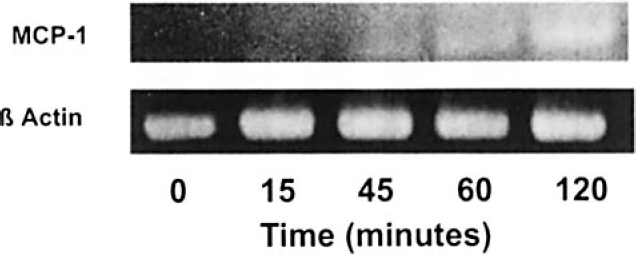
Kinetics of MCP-1 mRNA expression after stimulation with heat-killed Mh. Shown are the kinetics of MCP-1 mRNA expression using qualitative RT-PCR. A549 cells were treated with heat-killed Mh and total RNA isolated after 0 (untreated), 15, 45, 60, and 120 minutes. RT-PCRs were electrophoresed on 2% agarose gels and stained with ethidium bromide. RT-PCR products were compared with a standard DNA ladder.
Discussion
It is likely that the factors responsible for initiating the inflammatory component of BPD are complex and numerous. The presence of mycoplasmas in the lungs of premature infants who require mechanical ventilation must be considered a potential cofactor in the development of BPD, even when these microorganisms are considered to be of low virulence. This study provides further evidence of the pathologic potential of Mh in the lungs of preterm newborns by demonstrating that type II-like cells produce MCP-1 in response to viable and heat-inactivated Mh. Increases in lung MCP-1 concentrations could lead to recruitment to and activation of mononuclear cells in the alveolar space, which would lead to amplification of the inflammatory response. Active infection by Mh may not be required in this process because heat-killed Mh induced MCP-1 in A549 cells. These observations expand our earlier studies, which demonstrated that viable and heat-inactivated Mh induce IL-8 and ENA-78 production in type II-like cells. 14 Mh stimulates MCP-1 production by increasing MCP-1-specific mRNA. The induction of MCP-1 mRNA occurs within 2 hours of stimulation by heat-inactivated Mh. The kinetics of expression of MCP-1 mRNA is different than that of IL-8 mRNA, in which the maximal mRNA expression as detected by PRT-PCR occurred after 30 minutes of stimulation (unpublished observation, February 2000). The observed increase in MCP-1 mRNA is likely the result of increased gene transcription, although our experiments do not rule out the possibility of increased mRNA stability.
Induction of MCP-1 in A549 cells by Mh is not mediated by the production of IL-1α, IL-1β, or TNF-α. Incubation of A549 cells with IL-1ra and with neutralizing antibodies to these cytokines did not inhibit Mh-induced MCP-1 production. This is further supported by the observation that incubation with heat-killed Mh did not result in detectable concentrations of IL-1a, IL-1β, or TNF-α in conditioned media. This is in contrast to the IL-1a-mediated respiratory syncytial virus induction of intercellular adhesion molecule-1 in A549 cells. 25
Our experiments do not address the nature of the stimulatory factor. It is clearly not a secreted soluble factor, as is the case for Burkholderia cepacia induction of IL-8 by A549. 17 Furthermore, stimulation was not a result of LPS contamination or a LPS-like factor because polymyxin B, which blocks LPS stimulation, had no effect on MCP-1 induction by Mh. It is likely, however, that Mh-associated membrane lipoproteins mediate cytokine induction in A549 in a way similar to that described for other Mycoplasma species in mononuclear cells. 26
Mh infections may occur alone or with Uu. 20 Isolation of Mh in the lung of infants who developed BPD has been considered incidental and of less significance than that of Uu in the development of BPD. However, it is possible that the role of Mh in the development of BPD has been underestimated because of the high incidence of co-isolation of Uu. These results, combined with those of our previous studies, suggest that Mh alone has the potential to initiate a significant inflammatory response in preterm newborns.
The role of type II cells in the inflammatory responses of preterm infants is undetermined at present. Type II cells are found as early as the 20th week of gestation and constitute a significant percentage of lung cells at birth. At this time, the uninfected neonate has virtually no alveolar macrophages in the lung. 27 Thus, type II cell cytokine production could potentially initiate the pulmonary inflammatory response in the immediate newborn period. Additionally, production of MCP-1 by type II cells would help to further amplify the inflammatory response by recruitment and activation of alveolar macrophages.
The induction of MCP-1 by Mh in Type II cells may be of importance in the later phases of lung injury and subsequent development of fibrosis. Neutrophil influx mediated by IL-8 and ENA-78 is the hallmark of acute inflammatory processes, whereas chronic inflammation and fibrosis are characterized by a mononuclear cell infiltrate. Hence, the production of MCP-1 may be central to the pathogenesis of the fibrosis in BPD. In support of this is the observation that the concentration of MCP-1 is elevated in the lung lavage fluid from patients with the adult respiratory distress syndrome, and this increased MCP-1 correlates with the degree of lung injury in the late phase of this disease. 28 Furthermore, lung fluid concentration of MCP-1 is increased in diseases associated with pulmonary fibrosis found in both humans and animals.29–32 Lastly, evidence suggests that MCP-1 may directly increase collagen synthesis by fibroblasts. 33 We have recently demonstrated that tracheal aspirate fluid MCP-1 concentrations are increased in very low birth-weight infants who developed BPD compared with those who did not. 22 Increased concentrations of both MCP-1 and IL-8 were associated with the isolation of Uu and Mh in cultures of tracheal aspirates obtained in the first week of life. This suggests that mycoplasmas in the lungs and airways of preterm infants leads to a more robust inflammatory response, which in turn leads to increased lung injury and the development of BPD.
In summary, our data show that viable and heat-inactivated Mh can induce MCP-1 gene expression and protein production in type II-like cells in vitro. These data suggest that type II cell-mediated cytokine induction by these organisms contributes to the pulmonary inflammatory response that underlies the development of BPD in very low birth-weight infants.