
Abstract
This study reports one case of Kabuki syndrome (KS) accompanied by a new mutation in the KMT2D gene and concurrent congenital pulmonary airway malformation (CPAM). After birth, the child demonstrated delayed physical growth and neurological, psychological, and intellectual development. Notable facial features included arched eyebrows and elongated eyelids toward the lower side. Chest computed tomography revealed pulmonary airway malformation. Whole-exome high-throughput sequencing revealed a novel heterozygous missense mutation in the KMT2D gene, Exon38c.10481C>T (p.Pro3494Leu), a possible pathogenic mutation that has not been previously reported. Both parents were wild-type. The clinical manifestations of KS are complex; however, cases with concomitant CPAM have rarely been reported. This study elucidates the mutation spectrum of the KS gene and the clinical characteristics of KS patients.
Introduction
Kabuki syndrome (Niikawa–Kuroki syndrome, Kabuki make-up syndrome, KS, OMIM #147920) is a rare disorder characterized by distinctive facial features resembling those of Japanese Kabuki individuals, along with multisystem impairments such as growth retardation, skeletal anomalies, congenital visceral malformations, abnormal dermatoglyphics, and intellectual disability, 1 imposing a substantial burden on both society and affected families.
With the rapid advancement of second-generation sequencing technology, the pathogenic genes associated with KS were elucidated by 2010. 2 Currently, KMT2D and KDM6A are considered the main pathogenic genes of KS. About 80% of patients exhibit mutations in the KMT2D gene, which are classified as KS1 type and are inherited in an autosomal dominant manner. About 6%–10% of patients have mutations in the KDM6A gene, which is classified as the KS2 type and is inherited in an X-linked dominant manner.2–4 Mutations in the KMT2D gene lead to downregulation of KMT2D protein expression, which subsequently affects histone H3 lysine 4 (H3K4) methylation and disrupts early embryonic development. 5 Different mutation types in KMT2D contribute to phenotypic variability among children with KS. 6 To date, there have been no reports or mechanistic studies on KMT2D mutations associated with pulmonary airway malformations. This study presents a case of a missense mutation in the KMT2D gene co-occurring with pulmonary airway malformation, thereby enriching the spectrum of known KS-related gene mutations.
Methods
Objective
To assess the clinical characteristics, gene sequencing results, diagnosis, and treatment of secondary pulmonary infection in a child with Kabuki syndrome (KS) with a new KMT2D gene mutation and possible congenital pulmonary airway malformation (CPAM).
Genetic testing methods
Peripheral venous blood samples were collected from the proband and both parents. Genetic analysis was performed using whole-exome sequencing (WES) employing high-throughput sequencing technology by Berry Genomics (China). Suspected dynamic mutation findings identified by WES were further analyzed by Sanger sequencing. Additionally, Sanger sequencing validation was conducted on the corresponding genetic loci of the proband’s parents.
Results
Clinical data
The male patient, aged 7 years and 6 months, was admitted to our department with symptoms of coughing for 2 weeks and fever for 5 days. The birth history revealed that the child was G1P1, born prematurely at 35+3 weeks of gestation, and he weighed 2 kg at birth. There were no complications, such as birth injury or asphyxia. Fetal ultrasonography performed at 23 weeks of gestation suggested a possible congenital pulmonary airway malformation. Developmental history: The patient could hold his head steady at 5 months, sit independently at 8 months, and walk independently at 17 months. Currently, he is capable of gross motor activities such as jogging and jumping with both feet, but is unable to perform fine motor tasks such as holding a pen or buttoning clothes. He can only respond to simple questions with slow speech rates and imprecise pronunciation. Intellectual development was delayed, with a Wechsler scale IQ score of 59 at 7 years of age. Family history: Both parents were healthy and non-consanguineous. He had a 2-year-old younger brother who was healthy. There was no family history of genetic diseases. A pedigree chart is shown in Figure 1.
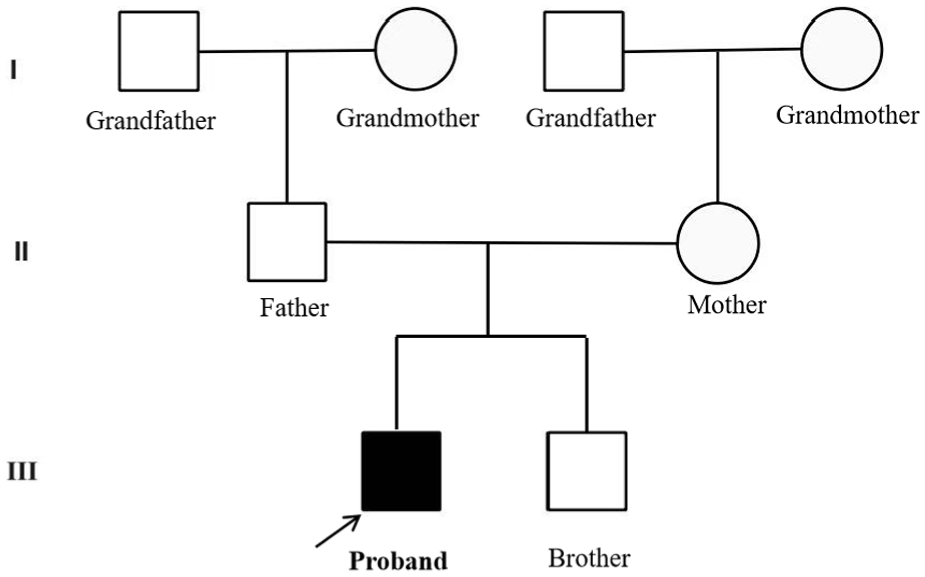
The pedigree chart indicates that the proband is the affected child, with no immediate family members having similar conditions.
The physical examination results were as follows: height 115 cm (−3 SD to −2 SD), weight 22.7 kg (−1 SD to median), head circumference 46 cm (<−3 SD), arched eyebrows, downward-slanting palpebral fissures, decreased breath sounds in the left lung, fine moist rales audible over the left lung, regular heart rhythm, normal heart sounds, no abnormalities in the appearance of the anus and external genitalia, bilateral single transverse palmar creases, and fetal pads of the fingertips.
Laboratory examination revealed the following values: white blood cells, 48 × 109/L; hemoglobin, 137 g/L; platelets, 354 × 109/L; neutrophils, 90.2%; high-sensitivity C-reactive protein, 112.87 mg/L; and real-time fluorescence quantitative PCR of bronchoalveolar lavage fluid for Mycoplasma pneumoniae: DNA< 2.5 × 103 copies/mL. The imaging findings were as follows: plain chest computed tomography (CT) revealed left lung pneumonia, possible bronchiectasis in the upper left lung, and varying sizes of cavity-like changes (Figure 2(a)).
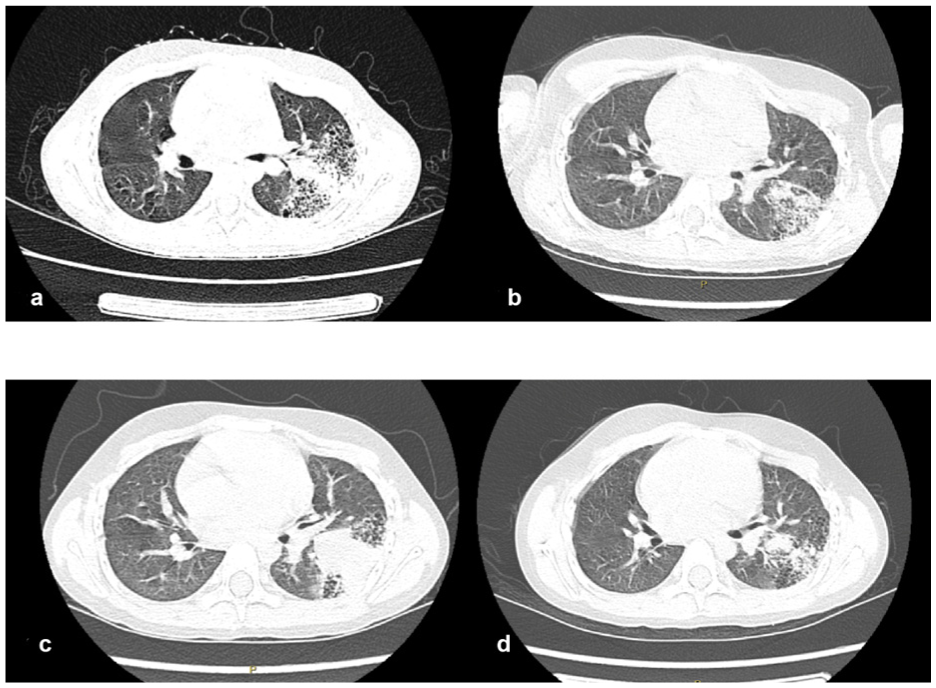
(a) demonstrates the chest computed tomography (CT) scan results from the first admission, exhibiting pneumonia in the left lung with varying sizes of cavity-like changes and possible cystic adenoma-like deformities; (b) presents the results of a chest CT plain scan after the first bronchoalveolar lavage and anti-infection treatment performed under fiberoptic bronchoscopy, indicating that the lung inflammation has improved compared to the initial scan; (c) is the high-resolution plain CT scan of the chest taken upon readmission, showing that the left lung inflammation has worsened than before; After the second round of bronchoalveolar lavage and anti-infection treatment under fiberoptic bronchoscopy, the chest CT plain scan; (d) was re-evaluated, demonstrating a significant reduction in lung inflammation compared to before.
Diagnosis and treatment process: The patient underwent fiberoptic bronchoscopy, and bronchoalveolar lavage was performed with normal saline in the right upper lobe, right middle lobe, right lower lobe, and left lower lobe bronchus. The lavage fluid was subjected to Mycoplasma DNA detection and bacterial culture. The microscopic examination revealed endobronchial inflammation, bronchial stenosis, and bronchial softening (mild). The child was treated using intravenous infusions of azithromycin, cefoperazone sulbactam, and methylprednisolone. These interventions alleviated the child’s clinical symptoms, and a follow-up chest CT indicated that the degree of inflammation had decreased (Figure 2(b)). The patient was discharged after hospitalization for 10 days.
Four days after discharge, the patient was readmitted due to “fever for 2 days”. Compared with previous scans, a high-resolution chest CT scan revealed that the inflammation in the left lung had progressed (Figure 2(c)). The child received intravenous infusions of azithromycin and linezolid for anti-infection, followed by a second fiberoptic bronchoscopy and bronchoalveolar lavage. A follow-up CT scan revealed that inflammation had decreased compared with that before hospitalization (Figure 2(d)), and the patient was discharged after hospitalization for 10 days. During our outpatient follow-up, we scheduled a lung tissue biopsy at the Department of Cardiothoracic Surgery to confirm any pathological changes.
Genetic testing results
A heterozygous missense mutation was observed in the KMT2D gene c.10481C>T (p.P3494L), which has not been reported to date. Neither parent carried a mutation at this locus, indicating a new mutation (Figure 3). According to the guidelines set forth for the application of guideline standards by the American Society of Medical Genetics and Genomics and the recommendations of the ClinGen Sequence Variation Interpretation Expert Group, this mutation is potentially pathogenic.7,8 The pathogenicity of the mutation site was assessed using the online software MutationTaster (http://www.MutationTaster.org/), which indicates predictive pathogenicity. According to the ClinVar database and the Human Gene Mutation Database, this variant site has not yet been included.
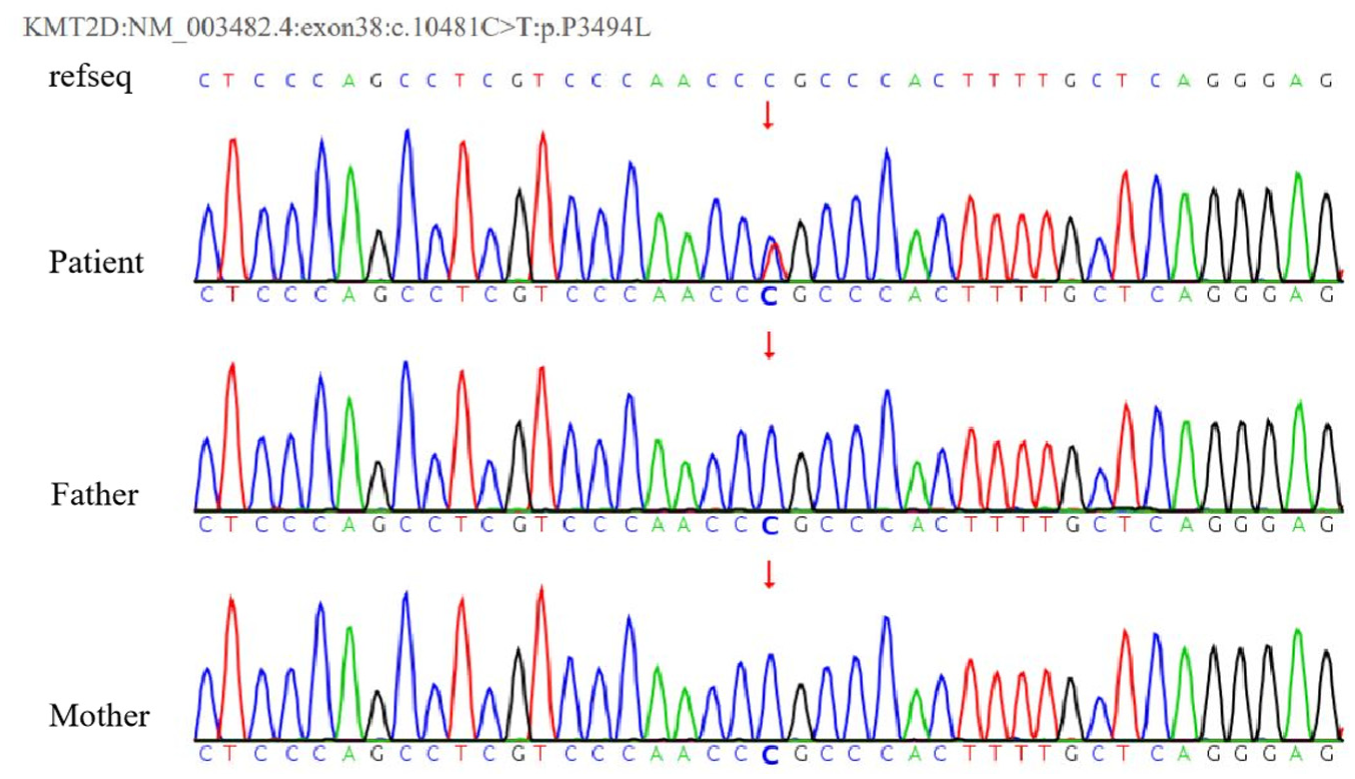
The KMT2D gene sequencing results of the patient and his parents demonstrate that the patient has a heterozygous mutation in the KMT2D gene, specifically in exon 38: c.10481C>T (p.P3494L), whereas neither the father nor the mother carries the mutation.
Discussion
According to the diagnostic criteria for KS established in the 2018 International Diagnostic Consensus, 9 the patient in this case presented with developmental delay, intellectual disability, and characteristics such as elongated eye fissures, arched eyebrows, and microcephaly. Genetic testing suggested the presence of pathogenic variations in the KMT2D gene, thus confirming the diagnosis of KS in this patient.
Recent research has focused on the molecular genetic mechanism of KS and the relationships between different types of gene mutations and clinical phenotypes. The KMT2D gene is located on chromosome 12q13.12, having a size of 36.3 kb, and contains 54 exons. It encodes the histone lysine N-methyltransferase 2D, which comprises 5537 amino acids. It belongs to the human histone transferase superfamily and mainly catalyzes the methylation of the fourth lysine site of histone H3 (H3K4). It is widely expressed in adult tissues and is critical for early embryonic development. Another major pathogenic gene is the KDM6A gene, located on chromosome Xp11.3, which contains 29 exons and encodes a histone demethylase comprising 1401 amino acids. Its primary function is to catalyze the demethylation of histone H3 lysine site 27 (H3K27), interact with the KMT2D protein, and jointly participate in the regulation of the histone methylation process.5,10
Histone methylation is one of the main epigenetic mechanisms involved in regulating gene expression. Methylation of H3K4 is used by cells to label promoters and enhancers, playing a critical role in regulating development, differentiation, metabolism, and tumor suppression. 5 The KMT2D protein performs its enzymatic function by catalyzing H3K4 methylation, which primarily depends on the C-terminal conserved SET domain N, the terminal PHD domain, the HMG group, and two FYRC/FYRN regions rich in the FY structure. Among these domains, the SET domain ensures enzyme activity and structural stability, whereas the PHD domain recognizes histone lysine sites on nucleosomes and participates in the methylation of H3K4.
Variations in gene mutations can lead to functional abnormalities in different protein domains, resulting in varying clinical characteristics among children with KS.6,11,12 In addition to having special facial features, developmental delays, and intellectual disabilities, these patients may also experience hearing loss or impairment, congenital heart disease (CHD), epilepsy, congenital megacolon, biliary atresia, ectopic kidney, horseshoe kidney, Hashimoto’s thyroiditis, autoimmune hemolysis, idiopathic hemolysis, and other multisystem impairments.
Mutations in the KMT2D/KDM6A genes exert global effects on the growth and development of craniofacial, visceral, neural, and musculoskeletal tissues. Previous studies have described certain distinctions in the clinical phenotypes associated with KMT2D and KDM6A gene mutations. For example, short stature is observed in up to 70% of KS patients, with a significantly higher prevalence among those carrying KMT2D variants than among noncarriers. 13 Deletions in KMT2D may lead to impaired synthesis of the protein’s N-terminal region, resulting in more pronounced developmental delays. 14 CHD is present in up to 80% of KS patients with KMT2D variants, with coarctation of the aorta and ventricular septal defects being the most common types. 15 A mouse model study indicated that KMT2D deletion reduces H3K4 methyltransferase expression at enhancer and promoter regions of genes involved in cardiac development, subsequently downregulating the expression of genes related to ion transport and the cell cycle. 16 Neuropsychological symptoms, including learning disabilities, reduced IQ, impaired adaptive skills, autism-like behaviors, and psychopathological manifestations such as anxiety, phobias, unusual behaviors, and emotional dysregulation, are also more frequently observed in patients with KMT2D variants. 17
Reports on the relationships between different mutation types and locations of the KMT2D gene and clinical phenotypes are scarce. A review involving 1,174 cases with pathogenic or likely pathogenic variants in the KMT2D gene indicated that the majority of the mutations were truncating (20.5% nonsense mutations and 11.7% frameshift mutations), which may lead to loss of SET domain activity and consequent protein dysfunction. Missense mutations accounted for 13.9%, followed by small deletions (8.1%) and splice-site variants (5.6%). Variants of the KMT2D gene occurred more frequently in exons 39, 48, 31, 34, 11, and 106.
It has been reported that missense mutations at specific sites in exons 38 or 39 of the KMT2D gene, which affect highly conserved amino acid residues, may lead to another syndrome, charge syndrome. 18 This disorder is characterized by branchial arch anomalies, ear malformations, eustachian tube atresia, lacrimal duct defects, abnormal nipple development, thyroid abnormalities, growth retardation, and immune system dysfunction. The present case did not exhibit typical features such as branchial arch anomalies, ear malformations, or eustachian tube atresia. In the literature, seven reported cases of Charge syndrome caused by KMT2D gene missense mutations presented no significant intellectual disability, whereas intellectual disability was present in our case.
This study reports a case of Kabuki syndrome with a KMT2D missense mutation co-occurring with CAMP, which was previously known as congenital cystadenoma-like malformation. CAMPs are characterized by abnormal alveolar structure, excessive proliferation and dilation of terminal bronchioles, and polycystic proliferation of lung tissues. Prenatal ultrasound screening is performed at 18–22 weeks of pregnancy to detect children with CPAM. Chest CT is the gold standard technique for studying CPAM19,20 and is considered the critical modality for the postpartum diagnosis of CPAM, with a sensitivity of 100%. The specific pathogenesis of CPAM is ambiguous, and abnormal gene expression of Sox2, Nkx2, Hoxb5, FABP7, PDGF-B, KRAS, and other genes may be related to the occurrence of CPAM, as suggested by some animal models and human studies of lung tissue resection.21, 22 The KMT2D and KDM6A genes, which are KS-related genes, are involved in histone methylation and demethylation. These processes are crucial for early embryonic development and differentiation. KDM6A gene mutations have been reported in patients with CPAM; however, no typical clinical manifestations of KS were observed in this case. 23 Currently, KMT2D gene mutations have not been reported in patients with CPAM. In the case of KS, a new heterozygous mutation in the KMT2D gene was detected, and chest CT demonstrated CPAM, indicating that the Exon38c.10481C>T (p.Pro3494Leu) mutation in the KMT2D gene may be related to CPAM occurrence. However, the specific pathogenic mechanism remains to be further explored.
In summary, KS has similar special facial features; however, significant differences in clinical phenotype characteristics exist among different systems. This poses challenges for pediatricians in the early diagnosis of this disease. Thus, whole-exome gene testing should be performed as early as possible for individuals suspected of having KS to confirm the diagnosis. Currently, there is a lack of specific treatment measures. Therefore, it is crucial to regularly follow up and evaluate the growth and development level, as well as the functions of various organs in the child’s system, to prevent complications and improve prognosis. The course of the disease is prolonged when children with KS are complicated with secondary pulmonary infection caused by CPAM, and a full course of anti-infection treatment should be administered based on the pathogen results. Fiberoptic bronchoscopy examination and lavage treatment should be conducted if needed.
Conclusions
This study reports the clinical manifestations of a KS case with a de novo KMT2D gene mutation. The KMT2D Exon38 c.10481C>T (p.Pro3494Leu) mutation may be associated with the occurrence of CPAM; however, the underlying pathogenic mechanism remains to be further elucidated.
Footnotes
Acknowledgements
We sincerely thank the patient and his parents for their cooperation in this study.
Ethical considerations
Not applicable.
Consent to participate
Informed consent was obtained from all individual participants.
Consent for publication
Not applicable
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Special fund projects for the research of intractable and rare diseases (2024YNHJ15).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.