
Abstract

Comparison Of Platelet-Activating Factor Levels In Infants With Necrotizing Enterocolitis And A Neonatal Pig Model.
S. Piecuch, G. Condemi, P. Dzakpasu, G. Valencia, E. Kornecki, P.M. Gootman, S. Rabinowitz (intr. by K. Bromberg), Department of Pediatrics, State University of New York - Health Sciences Center, Brooklyn, NY.
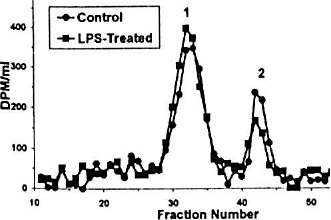
Lipopolysaccharide-Induced Inflammation Results In Loss Of Glycosaminoglycans.
MS Salat, HS Dweck and L Rosenfeld. Neonatal Research Laboratory, Westchester Medical Center, New York Medical College, Valhalla, NY.
Necrotizing enterocolitis (NEC) is an acute inflammatory disease of newboms. In vitro studies by our group have shown that products of nitric oxide degrade extracellular matrix glycosaminoglycans (GAGs), such as heparan sulfate (HS) or chondroitin sulfates (CS). The purpose of this study is to determine whether these GAGs are degraded in intestines of rats treated with lipopolysaccharide (LPS).
Zonula Occludens Toxin (Zot) Inhibits Intracellular Trafficking Of Luciferase Dna In Susceptible Cells.
T. L. Watts, J. Malone, R. Malone, A. Fasano, Center for vaccine Development and Division of Pathology, University of Maryland, Baltimore, MD.
Zonula occludens toxin (Zot) is a protein elaborated by Vibrio Cholerae that modulates the opening of intercellular tight junctions (tj). The mechanism by which Zot modulates tj involves rearrangement of the cytoskeleton with resultant polymerization of actin fibers via a PKC dependent pathway. Thus, Zot promotes an increase in tissue permeability and a reduction in transepithelial resistance by opening tj. Preliminary data suggests that Zot is a microtubule binding protein capable of modulating normal intracellular trafficking patterns. Using a luciferase trasnfection assay, various cell lines were trans-fected with luciferase DNA in the presence or absence of Zot. In addition, cells were exposed to Zot either before or after luciferase transfection.
Three general phenotypes were observed. 410.4 cells showed a reduction in DNA expression either after pre- or post-treatment with Zot. DU145, NIH 3T3, and Cos-7 cells lacked DNA expression only when Zot was administered after DNA transfection. In cells known to be deficient for the Zot receptor (Iva, T84) there was no observable effect on luciferase expression in the presence of Zot. These obser-vations have led to the conclusion that Zot may inhibit DNA expression due to its ability to modulate intracellular trafficking.
An Unusual Presentation Of Extra-Hepatic Biliary Atresia.
J Rosado, A.R. DeFelice, F Zapata, R Ramon, N Sharma, and P.G. Kazlow. Department of Pediatrics, Columbia University, New York, NY.
It is believed that extra-hepatic biliary atresia (EHBA) is typically not found in premature infants. In addition, children with EHBA classically present with light-colored, or acholic, stools. The patient, a 32 week gestation, twin A, was delivered via caesarean section. Birth weight was 1170 g. Apgars were 7 and 8. The patient was fed enterally and never received hyperalimentation. The neonatal course was complicated by sepsis, which responded to antibiotic treatment. The infant was noted to have conjugated hyperbilirubinemia (6.0/2.3mg/dl) on day 2 of life. The direct bilirubin peaked at a level of 9.3/3.7 mg/dl, on day 5 of life, then began to decrease. However, the bilirubin then reached a plateau of 9.3/2.0 mg/dl. Liver function tests were elevated, including alkaline phosphatase 231 u/l, AST 55 u/l, ALT 53 u/l and GGT P 2074 u/l. Work-up for infectious and metabolic etiologies was negative. The patient was then placed on Phenobarbital. An initial DISIDA scan revealed no excretion of isotope into the duodenum. During this entire time, the child continued to produce stools that were greenish-yellow in color. The child was then placed on Actigal, and a repeat DISIDA scan again showed no excretion. A percutaneous liver biopsy, at age 11 weeks, revealed bile ductule proliferation, inflammation, and mild fibrosis. The child was taken to the operating room, where he underwent a Kasai (hepatoportoenterostomy) procedure. This resulted in a fall of the serum bilirubin and an improvement of the liver function tests. EHBA is the result of a progressive, inflammatory process that leads to the obliteration of the biliary tract. Cases in still-births, or in premature infants, are rare. Stools are generally acholic at the time of presentation. The etiology of this condition is unknown. Many theories have been proposed, including infectious, ischemic, immunologic and toxic causes. Our patient was intriguing in that he was premature and maintained pigmented stools throughout the course of his work-up. The Kasai procedure is most successful when performed before 60 days of age. We therefore suggest an aggressive work-up of all infants with neonatal jaundice, even those born prematurely and having pigmented stools, to rule out EHBA as a diagnostic entity.
Epstein-Barr Virus Hepatitis As A Precursor To Scleroderma In An Adolescent.
E Treskova, I Shendrik*, N Galeano, A.R. Depelice, and P.G. Kazlow. Departments of Pediatrics and Pathology*, Columbia University, New York, NY.
Scleroderma has classically been regarded as a dermatological condition. Recent studies have described multi-system involvement, particularly in the gastrointestinal tract. A 14 y.o. female presented to our hospital with complaints of fatigue, dysuria, swollen feet and a 20 lb weight gain. Physical examination was notable for marked bilateral pitting edema of the lower extremities. Laboratory data revealed a normal CBC. There was hypoalbuminemia (2.8 g/dl) and elevation of the liver enzymes (AST 113 u/l; ALT 148 u/l). Alkaline phosphatase and GGT were normal. An autoimmune work-up, including ANA, anti-DNA, anti-cardiolipin antibodies, anti-mitochondrial antibodies, was negative. Smooth muscle antibody titers were positive at 1:80. Serology for hepatitis A, B, and C was negative. Abdominal ultrasound revealed splenomegaly, with a normal-appearing liver. The patient experienced an initial clinical improvement but soon became worse. At reevaluation 3 months later, she had developed hepatomegaly and elevation of all liver function tests (AST 356 u/l; ALT 58 u/l; GGT 202 u/l). EBV-VCA titers, which had initially been negative, were now increased to 1:2500. The patient underwent a percutaneous liver biopsy, which revealed diffuse sinusoidal lymphocytic infiltration consistent with EBV infection. The patient again experienced a clinical remission with the use of vigorous diuretic therapy. Three months later, she presented again, with stiffness of the carpal, metacarpal and tarsal joints. She had also developed areas of patchy hypo- and hyperpigmentation of her skin. There was also the onset of Raynaud's phenomenon and dyspnea upon exertion. At that time, a diagnosis of scleroderma was made. She was treated with Prednisone and 6-MP and experienced significant improvement, both clinically and in all biochemical parameters. Scleroderma is a diagnosis that is only infrequently made in the pediatric age group. There is a great deal of speculation as to the role of infectious agents, particularly the Epstein-Barr Virus, in auto-immune disease. The Epstein-Barr nuclear antigen-1 has a highly antigenic glycine-alanine region which cross reacts with similar epitopes in many human proteins. The clinician should be alerted to look for possible auto-immune disease in the patient who presents with that which appears to be multi-system EBV infection.
Neonatal Cholestasis And Urinary Thiobarbituric Acid Reactive Substance (Utbars) Levels.
Kazimierz Watorek, Richard Strauss, Gisela Wirz, Mark Hiatt, Thomas Hegyi UMDNJ-RWJ Medical School, St. Peter's Medical Center, Div of Neonatology, and Dept of Environmental and Community Medicine, N Brunswick, NJ.
Neonatal cholestasis (Direct bilirubin level, (DBL) >2mg/dl on day >10) was associated with increased levels of uTBARS in a group of preterm infants. Urine samples from 10 cholestaic infants (BW 1034±667g; GA 27.4±4.7wks) and 27 controls (BW 1306±679g; GA 29.4±4.1wks) were collected on 39.4±35.6 days of life and frozen at -70°F for later examination. The pH adjusted sample was combined with TBA, heated, and analyzed by spectrophotometric analysis. The groups were similar with respect to Apgar scores, oxygen requirement, arterial oxygen content, and serum bilirubin levels during the first week of life. uTBARS excretion levels (ng/mg of creatinine) are shown below:
p<0.05
p=0.1
Multiple regression analysis revealed significant relationships between DBL and oxygen requirement, maximal total serum bilirubin concentration, and birth weight. Of note, there were no relationships noted between DBL and intravenous nutrition data. uTBARS levels were not associated with any of these variables. In these infants, cholestasis was associated with abnormal liver function tests, and with a non significant increase in uTBARS, products of lipid peroxidation, suggesting a role for reactive oxygen substances in liver damage.
Linkage Disequilibrium Analysis Of The Gaucher Disease N370S Mutation.
1GA Diaz, 1BD Gelb, 2N. Risch, 3T. Nygaard, 4I. Maire, 5L. Poenaru, 5C. Caillaud, 6C. Sa Miranda, 6O. Amaral, 1PK. Mistry and 1RJ. Desnick. 1Department of Human Genetics, Mount Sinai School of Medicine, New York, NY; 2Department of Genetics, Stanford University, Stanford, CA; 3Department of Neuroscience, UMDNJ-New Jersey Medical School, Newark, NJ; 4Hopital Debrousse, Lyon, France; 5Laboratoire de Genetique, Hopital Cochin, Paris, France; 6Institute de Genetica Medica, Porto, Portugal.
Gaucher disease, a lysosomal storage disease caused by glucocerebrosidase (GBA) deficiency, is far more common in the Ashkenazi Jewish (AJ) population, for reasons which remain controversial. Previous work with GBA polymorphisms suggested that the commonest mutation, N370S, arose from a founder in the AJ population. To investigate the genetic history of this disorder, a 9.2-cM region containing the GBA locus was finely mapped with 10 short tandem repeat markers which were then used for genotyping AJ (n=140) and non-AJ Portuguese (n=27), and French (n=19) chromosomes carrying N370S mutations. Marker allele frequencies were generated for N370S and control chromosomes from each population. Greater than 92% of the AJ N370S haplotypes were conserved at three of the four central markers, indicating the existence of a common founder. The AJ N370S haplotype was observed frequently in the French and Portuguese populations, although about 26% and 33%, respectively, had divergent haplotypes, indicating recurrent N370S mutation events. The linkage disequilibrium parameter, δ, was calculated for the marker loci. The results for the AJ N370S were consistent with a more ancient coalescence for the GBA mutation than for the common AJ idiopathic torsion dystonia mutation which had previously been dated back ~17 generations (~1650). French and Portuguese N370S chromosomes carrying the central AJ N370S haplotype had less overall linkage disequilibrium at loci flanking the conserved core haplotype than did the AJ N370S chromosomes. These findings disprove the hypothesis that the N370S mutation arose in the Ashkenazi population and then was bred into the non-Jewish European populations by admixture. Rather, the AJ N370S was ancient in Europe and rose to high prevalence after its introduction into the AJ population. The finding that AJ and European non-AJ N370S chromosomes share a common origin do not support models positing heterozygote advantage as a mechanism to explain the high frequency of the N370S mutation in the AJ population.
Prevalence Of Celiac Disease Among First And Second Digree Relatives In The U.S.A.
I. Berti, K. Horvat, T. Not, A. Fasano. Division of Pediatric GI and Nutrition, University of Maryland School of Medicine, Baltimore, MD U.S.A.
1. Scand J Gastroenterol 1998:33:494-498.