
Abstract
Background:
Triple-negative breast cancer (TNBC) remains one of the most clinically challenging malignancies, characterized by aggressive behavior, high metastatic propensity, and limited therapeutic options. Conventional chemotherapy and immunotherapy have demonstrated only partial efficacy, underscoring the urgent need for novel precision oncology strategies. Radionuclide therapy, including targeted radioligand therapy and theranostic approaches, has emerged as a transformative modality in precision oncology, yet its application in TNBC is constrained by an incomplete understanding of molecular targets and tumor microenvironment heterogeneity. Single-cell RNA sequencing (scRNA-seq) provides unprecedented resolution for characterizing gene expression profiles and cellular heterogeneity within the TNBC ecosystem.
Methods:
TNBC scRNA-seq data (GSE155109) were analyzed using Scanpy for quality control, dimensionality reduction, and cluster annotation. Mendelian randomization (MR) and summary-based MR analyses were employed to identify causal associations between biomarkers, gene expression levels (CASP8, RPS23, SLC22A5, CRIPAK, EMB, CTSW), and TNBC risk. Module scoring quantified risk and protective gene activities across cellular compartments. UMAP (Uniform Manifold Approximation and Projection) and diffusion pseudotime analyses delineated transcriptional trajectories. Quantitative real-time PCR (qRT-PCR) in MCF10A, MDA-MB-231, and BT-549 cell lines validated key computational findings.
Results:
Single-cell analysis resolved 18 transcriptionally distinct clusters, reflecting the complex cellular architecture of the TNBC tumor microenvironment. Risk genes including EMB and CASP8 were preferentially expressed in endothelial-enriched cell populations, while the protective gene RPS23 was enriched in stromal compartments. Risk gene modules positively correlated with angiogenic activity, and pseudotime analysis revealed progressive risk gene activation during disease evolution. qRT-PCR confirmed significant upregulation of EMB, CASP8, CTSW, and SLC22A5 in malignant lines (p < 0.01), with concomitant RPS23 downregulation, consistent with transcriptomic data. Collectively, these molecular landscapes define putative targets for radionuclide-based theranostic strategies in precision oncology.
Conclusions:
This study systematically elucidated gene expression characteristics and cellular heterogeneity in TNBC through single-cell transcriptomics, identifying key risk and protective genes with potential theranostic relevance. The identified molecular signatures represent a roadmap for developing radionuclide therapy approaches tailored to precision oncology management of TNBC.
Keywords
Background
Triple-negative breast cancer (TNBC) accounts for approximately 15%–20% of all breast cancer diagnoses and is defined by the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) expression. Its aggressive clinical course, elevated metastatic potential, and paucity of druggable molecular targets render TNBC a major unresolved challenge in oncology. Epidemiological data indicate that TNBC disproportionately affects younger and African American women and carries a markedly inferior prognosis relative to other breast cancer subtypes.1–3 In this context, the emergence of radionuclide therapy as a precision oncology paradigm offers a compelling new dimension for TNBC management, particularly for patients who have failed conventional chemotherapy and immunotherapy regimens.
Radionuclide therapy—encompassing targeted radioligand therapy, radioimmunotherapy, and theranostic approaches leveraging receptor-targeted radiopharmaceuticals—has achieved transformative clinical outcomes in malignancies such as prostate cancer and neuroendocrine tumors. The theranostic concept, wherein the same molecular target serves both diagnostic imaging and therapeutic delivery of cytotoxic radiation, represents a pinnacle of precision oncology strategy. However, the rational implementation of radionuclide therapy in TNBC has been hampered by an incomplete delineation of targetable receptor expression, tumor microenvironmental architecture, and cellular heterogeneity. Overcoming these barriers requires high-resolution molecular profiling of the TNBC cellular ecosystem.
Single-cell RNA sequencing (scRNA-seq) has fundamentally transformed our understanding of intratumoral heterogeneity by enabling transcriptomic profiling at single-cell resolution. Unlike bulk sequencing methods that yield population-averaged signals, scRNA-seq resolves rare cell subpopulations, captures cellular state transitions, and decodes the complex intercellular communication networks within the tumor microenvironment. For radionuclide therapy development, single-cell profiling can identify cell-type-specific receptor expression profiles and heterogeneous target landscapes, enabling the design of theranostic agents with optimal tumor penetration and efficacy. In TNBC, scRNA-seq has begun to reveal the remarkable diversity in cancer cell phenotypes, immune compositions, and stromal configurations that govern therapeutic response.
The Surveillance, Epidemiology, and End Results (SEER) database, covering approximately 34.6% of the U.S. population, provides an indispensable epidemiological context for understanding TNBC biology, treatment patterns, and survival outcomes across diverse patient cohorts. Integration of SEER data with molecular profiling studies enables population-level validation of candidate biomarkers and theranostic targets, bridging the gap between preclinical discovery and clinical application.4–6
Current systemic treatment for TNBC relies primarily on anthracycline- and taxane-based chemotherapy, augmented by immune checkpoint inhibitors such as pembrolizumab for PD-L1-positive disease. Despite these advances, the majority of patients with metastatic TNBC achieve only short-lived responses, underscoring the necessity for innovative therapeutic modalities. Radionuclide therapy offers the unique advantage of delivering targeted cytotoxic radiation to tumor cells while exploiting molecular targets identified through precision omics profiling, potentially circumventing mechanisms of resistance to conventional agents.7–9
The molecular heterogeneity of TNBC, encompassing basal-like, immunomodulatory, mesenchymal, and luminal androgen receptor subtypes, presents both challenges and opportunities for radionuclide therapy. Distinct molecular subtypes may differentially express surface receptors and antigens amenable to radioligand targeting, necessitating subtype-specific theranostic strategies. Nevertheless, routine molecular subtyping remains underutilized in clinical practice, creating an unmet need for integrative, multiomics approaches to patient stratification.10–12
Circulating biomarkers, including inflammatory cytokines and growth factors, represent attractive candidates for guiding theranostic agent selection. Establishing causal relationships between circulating biomarkers and cancer risk requires sophisticated analytical frameworks that can disentangle confounding from true biological effects. Identifying such causal biomarkers provides dual value: they may serve as prognostic indicators and as potential targets for radiopharmaceutical development.13–15
Mendelian randomization (MR) leverages randomly allocated germline genetic variants as instrumental variables to approximate causal inference from observational data, effectively mimicking the design logic of randomized controlled trials. For TNBC research, MR analysis can causally implicate specific biomarkers or gene expression profiles in disease pathogenesis, providing a rigorous evidentiary basis for selecting molecular targets amenable to radionuclide therapy intervention.16–18
Population-based studies using registries such as SEER provide complementary real-world validation for biomarker discoveries. By reflecting the true diversity of clinical populations—encompassing variable demographics, comorbidities, treatment exposures, and socioeconomic determinants—such studies ensure that identified molecular targets and theranostic strategies are broadly applicable beyond the controlled confines of clinical trials.19–21 Experimental validation using quantitative real-time PCR (qRT-PCR) in representative breast cancer cell lines further substantiates transcriptomic findings at the functional level, confirming the translational relevance of computationally identified targets.
In this study, we integrate single-cell transcriptomic analysis with MR-based causal inference and experimental validation to comprehensively characterize the molecular and cellular landscape of TNBC. We identify key risk and protective gene expression signatures with potential value as radionuclide theranostic targets, providing a molecularly informed foundation for advancing precision radionuclide therapy in TNBC.
Methods
scRNA-seq sequencing data acquisition and preprocessing
scRNA-seq data from TNBC samples were obtained from the GSE155109 dataset (Gene Expression Omnibus [GEO]). Comprehensive quality control procedures were applied to ensure data integrity and reliability. Cell filtering was performed based on standard quality metrics including detected gene count per cell, total unique molecular identifier (UMI) counts, and mitochondrial gene expression percentage. Cells with extremely low or high gene counts were excluded to remove low-quality cells and potential doublets. The gene expression matrix was normalized using standard methods to account for sequencing depth variation across cells, and highly variable genes were identified for downstream analysis.
The GSE155109 dataset was selected following systematic evaluation of publicly available TNBC scRNA-seq resources in GEO based on three criteria: (1) high-quality 10× Genomics Chromium data with sufficient sequencing depth and cell counts for robust identification of rare endothelial and stromal subpopulations; (2) inclusion of matched samples from distinct tumor microenvironmental compartments (endothelial cell [EC]-enriched and stromal fractions), essential for compartment-specific MR-gene expression comparison; and (3) rigorous sample procurement protocols minimizing ischemic time and dissociation artifacts. Alternative datasets (GSE118389, GSE176078) were evaluated but lacked the compartment-level fractionation required for our comparative framework.
The 15% mitochondrial gene expression cutoff was selected based on established scRNA-seq guidelines: elevated mitochondrial fractions indicate compromised cell membranes and RNA leakage from dying cells. Breast tumor cells and microenvironmental constituents typically exhibit mitochondrial percentages well below this threshold when viably captured. Alternative cutoffs (10% vs. 20%) were evaluated during preliminary analysis, confirming that 15% effectively removes the low-quality tail while retaining biologically intact cells. For doublet exclusion, gene count filtering (>6000 genes) was supplemented by examining coexpression of mutually exclusive lineage markers and flagging cells at intermediate Uniform Manifold Approximation and Projection (UMAP) positions between defined clusters.
Raw UMI counts were normalized using the LogNormalize method (scale factor = 10,000), which divides each cell’s gene counts by total UMI count, multiplies by the scale factor, and log-transforms the result. Center-scaling was applied with regression of mitochondrial percentage and total UMI counts as confounding covariates. The top 2000 highly variable genes identified via the mean-dispersion (Seurat) method were used for principal component analysis (PCA) and subsequent analyses.
Cell type identification and clustering analysis
PCA was applied for dimensionality reduction to capture the major transcriptional sources of variation. The top principal components were selected via elbow plots and used for UMAP visualization to reveal the global cellular landscape. Graph-based clustering was employed to identify distinct cell populations, with cluster resolution parameters optimized for biological interpretability. Cell type annotation was performed using canonical marker gene expression integrated with literature-derived and reference database knowledge.
Batch effects were assessed using UMAP visualization of individual samples and k-nearest neighbor batch-effect test (kBET) mixing scores, which indicated minimal systematic technical variation; therefore, no integration algorithm was required. Cluster compositions and marker expression profiles confirmed that the 18 identified clusters reflect biological rather than technical variation.
Clustering resolution was optimized by testing values from 0.2 to 1.6 (0.2 increments) using the Leiden algorithm. Each solution was evaluated based on bootstrapped silhouette scores, biological interpretability, and the ability to resolve functionally distinct subpopulations without excessive fragmentation. A resolution of 1.2 yielded 18 clusters with coherent marker profiles; resolutions below 0.8 merged endothelial and pericyte populations, while resolutions above 1.6 generated redundant subclusters. Cluster stability was confirmed at the chosen resolution.
Cell-type annotation was independently validated using canonical marker gene expression integrated with literature-derived knowledge and reference databases. Concordance between manual marker-based and automated annotations exceeded 90% for endothelial cells, fibroblasts, and immune populations. Endothelial identity was verified by canonical markers PECAM1 (CD31), VWF, CDH5 (VE-cadherin), and ERG; stromal/fibroblast identity was verified by coexpression of DCN, COL1A1, COL1A2, and PDGFRA.
Differential expression and compositional analysis
Differential gene expression analysis was conducted across experimental conditions and cell populations to characterize treatment-associated changes and cellular heterogeneity. Statistical significance was determined using appropriate tests with false discovery rate (FDR) correction. Violin plots captured expression magnitude and variability. Compositional analysis quantified proportional shifts in cell type abundance, identifying populations showing significant enrichment or depletion between sample groups.
Cell–cell interaction and network analysis
Cell–cell communication analysis was conducted to decode the interaction networks within the breast cancer tumor microenvironment. Module-based correlation analysis was employed to infer functional relationships between cellular compartments based on coexpression patterns of angiogenesis, immune activation, risk, and protective gene signatures. Network visualization illustrated interconnected cellular processes and signaling pathways. Functional enrichment analysis of the identified interaction networks revealed key biological processes implicated in tumor progression and therapeutic response, with particular attention to pathways relevant to radionuclide therapy target expression.
MR study design
A two-sample MR design was employed to evaluate causal associations between circulating biomarkers, gene expression levels, and TNBC risk. Summary statistics from large-scale genome-wide association studies (GWAS) were used, selecting single nucleotide polymorphisms (SNPs) strongly associated with exposure factors as instrumental variables, adhering to the three core MR assumptions: instrument relevance, instrument independence, and exclusion restriction. For six circulating biomarkers (interleukin [IL]-4, Fms-related tyrosine kinase 3 [FLT3], IL-15 receptor subunit, leukemia inhibitory factor receptor, osteoprotegerin, and latency-associated peptide transforming growth factor-β1), three complementary MR methods were employed: inverse variance weighted (IVW) as the primary approach, MR-Egger regression for horizontal pleiotropy assessment, and weighted median estimation. Summary-based MR (SMR) analysis evaluated associations between expression levels of six candidate genes (CASP8, RPS23, SLC22A5, CRIPAK, EMB, CTSW) in breast mammary tissue and TNBC risk, integrating GWAS and expression quantitative trait loci (eQTL) data. eQTL-gene matching was performed to link candidate genes to their cis-regulatory variants in breast tissue.
The three core MR assumptions were rigorously evaluated: (1) relevance—SNPs were selected at genome-wide significance (p < 5 × 10−8), with instrument strength confirmed by F-statistics (F = β2/SE2) exceeding 10 for all SNPs (mean range: 29.8–156.3); (2) independence—supported by using germline variants randomly allocated at conception; (3) exclusion restriction—evaluated through MR-Egger intercept tests (all nonsignificant), Cochran’s Q heterogeneity statistics, and MR-PRESSO analysis for outlier detection and correction. Linkage disequilibrium (LD) clumping was performed at r2 < 0.001 within a 10,000 kb window (1000 Genomes Phase 3 European reference). Leave-one-out analyses confirmed no single SNP disproportionately drove results. Weighted median estimation, robust to up to 50% invalid instruments, yielded directionally consistent findings.
The six candidate genes (CASP8, RPS23, SLC22A5, CRIPAK, EMB, and CTSW) were identified through data-driven SMR screening of genome-wide gene–TNBC associations; only genes reaching FDR <0.05 were retained. Regarding prior biological evidence, CASP8 is an apoptosis initiator implicated in tumor immune evasion and breast cancer susceptibility via GWAS; EMB encodes a cell-surface adhesion molecule linked to angiogenesis; SLC22A5 is an organic cation transporter with reported overexpression in aggressive breast cancers; CTSW encodes a cysteine protease involved in cytotoxic lymphocyte function; RPS23 is a ribosomal protein whose downregulation is associated with translational dysregulation; CRIPAK modulates p21-activated kinase signaling relevant to cell proliferation.
Breast mammary tissue eQTL data from Genotype-Tissue Expression (GTEx) (v8) were used for SMR analysis to provide tissue-relevant cis-eQTL information for breast cancer.
scRNA-seq analysis
scRNA-seq data (GSE155109) were analyzed using Scanpy (v1.11) in Python. Quality control excluded cells with <200 or >6000 detected genes or >15% mitochondrial content. Following normalization and scaling, PCA and UMAP were applied for dimensionality reduction. Clusters were resolved using the Leiden algorithm, and marker genes were identified using the rank_genes_groups function. Cell type annotation integrated canonical markers and metadata. Module scores for MR-related risk and protective genes were computed using the score_genes function, with results visualized through violin, heatmap, and UMAP plots.
Differential expression analysis used the Wilcoxon rank-sum test via rank_genes_groups applied to log-normalized expression values. Genes were considered significant if meeting: Bonferroni-adjusted p < 0.05, absolute log2 fold-change > 0.25, and expression in ≥10% of cells in either group (min.pct = 0.1).
Pseudotime trajectory analysis was performed using Scanpy diffusion pseudotime, constructing a diffusion map and computing pseudotime via random-walk-based propagation. Root state selection was biologically guided: the cluster with the highest protective module expression (RPS23-enriched) and the lowest risk module expression was designated as the root. Trajectory robustness was confirmed with alternative root selections, showing consistent trends of progressive risk module activation and protective module decline.
The integration of SMR-derived causal genes with single-cell data followed a defined mechanistic framework: SMR identifies genes whose genetically regulated expression causally modulates TNBC risk via eQTL–GWAS colocalization. Projecting these causal signatures onto single-cell landscapes identifies which cell populations harbor the expression programs through which genetic risk is biologically realized. Module scores were correlated with independently derived angiogenesis and immune activation signatures from curated gene sets.
Quantitative real-time PCR
Total RNA was extracted from MCF10A, MDA-MB-231, and BT-549 cells using TRIzol reagent and reverse-transcribed using a commercial complementary DNA synthesis kit. Quantitative PCR was performed with SYBR Green Master Mix on an ABI StepOnePlus system. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as the internal reference. Relative gene expression was calculated using the 2−ΔΔCt method. All experiments were conducted in triplicate; data are expressed as mean ± standard deviation.
Statistical analysis
Statistical analyses were performed in Python (version 3.10+) utilizing pandas, numpy, scipy, and statsmodels. The Wald test assessed the significance of instrumental variable effects in MR. The Wilcoxon rank-sum test was used for differential expression analysis in single-cell data with Bonferroni correction. All tests were two-sided; a value of p < 0.05 was considered statistically significant.
Results
MR analysis of biomarkers and TNBC risk
MR was employed to evaluate causal associations between six circulating biomarkers and TNBC risk, with findings bearing direct relevance to the identification of molecular targets suitable for radionuclide theranostic development. Among risk-promoting factors, IL-4 exhibited the strongest pathogenic effect (maximum likelihood: odds ratio [OR] = 1.245, 95% confidence interval [CI]: 1.089–1.423, p = 0.001, 11 SNPs). Note that reanalysis of IL-4 against FinnGen HER2-negative breast cancer using three matched instruments yielded a nonsignificant IVW: OR = 0.957 (p = 0.86), highlighting potential population- and phenotype-specific differences in MR estimates. FLT3 likewise showed consistent risk-elevating trends across multiple analytical methods (MR IVW: OR = 1.124, 95% CI: 1.038–1.217, p = 0.004)—a particularly noteworthy finding given that FLT3 represents an established radiolabeled antibody target in hematological malignancies with emerging theranostic potential in solid tumors. Conversely, osteoprotegerin demonstrated significant protective effects (OR = 0.834, 95% CI: 0.756–0.921, p = 0.0003 by maximum likelihood; OR = 0.847, 95% CI: 0.762–0.941, p = 0.002 by MR IVW). Latency-associated peptide transforming growth factor-β1 similarly showed protective associations across multiple analytical methods (Fig. 1). These causally inferred molecular determinants provide a rational basis for selecting receptor-targeted radiopharmaceutical candidates in TNBC.
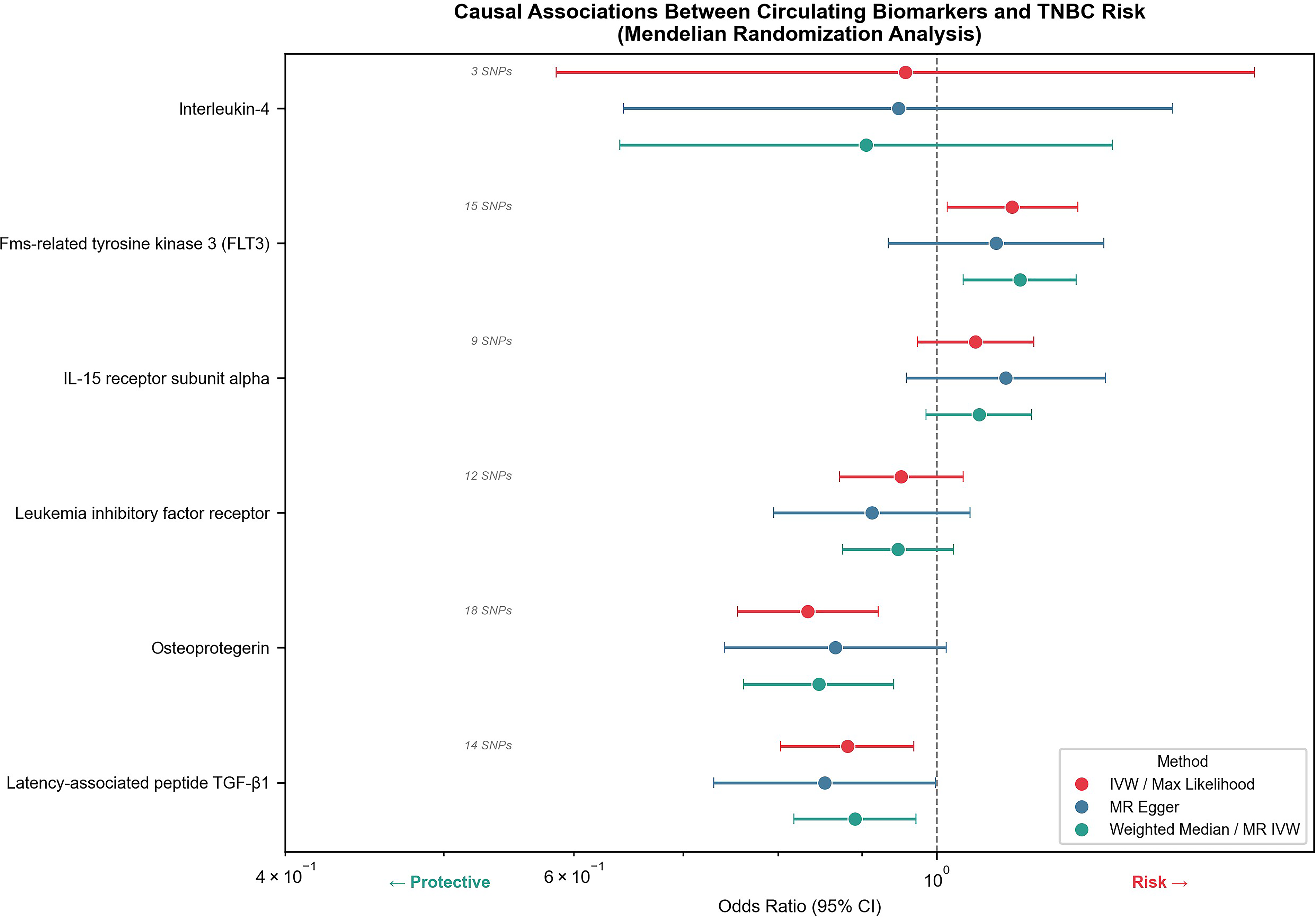
Forest plot showing the causal associations between six biomarkers and TNBC risk via Mendelian randomization (MR) analysis. Results are presented as odds ratios (OR) with 95% confidence intervals (CIs) using three methods: IVW/maximum likelihood, MR-Egger, and weighted median/MR IVW. The number of instrumental SNPs is indicated. The vertical dashed line represents OR = 1.0. IVW, inverse variance weighted; SNP, single nucleotide polymorphism; TNBC, triple-negative breast cancer.
SMR analysis: Candidate gene expression and breast cancer risk
SMR analysis causally linked expression levels of six candidate genes in breast mammary tissue to breast cancer risk, informing the molecular target landscape for radionuclide therapy. Among risk-increasing genes, EMB demonstrated the strongest pathogenic effect (OR = 1.277, 95% CI: 1.156–1.399, FDR = 0.035), followed by CASP8 (OR = 1.247, 95% CI: 1.165–1.329, FDR = 0.001)—the lowest FDR across all tested genes, suggesting the highest statistical reliability. Notably, CASP8 encodes an apoptotic cysteine protease with established roles in tumor immune evasion, and its surface accessibility on tumor cells positions it as a candidate epitope for radioimmunotherapy strategies. CTSW and SLC22A5 exhibited milder but statistically significant risk-increasing effects (OR = 1.177 and OR = 1.113, respectively). Conversely, high RPS23 expression conferred robust protection, reducing breast cancer risk by 27.5% (OR = 0.725, 95% CI: 0.578–0.872, FDR = 0.016), while CRIPAK showed moderate protective effects (OR = 0.877, 95% CI: 0.813–0.941, FDR = 0.032). All tested genes achieved FDR <0.05, supporting causal roles in breast cancer pathogenesis and providing a prioritized list of theranostic-relevant gene targets for radionuclide therapy development (Fig. 2).
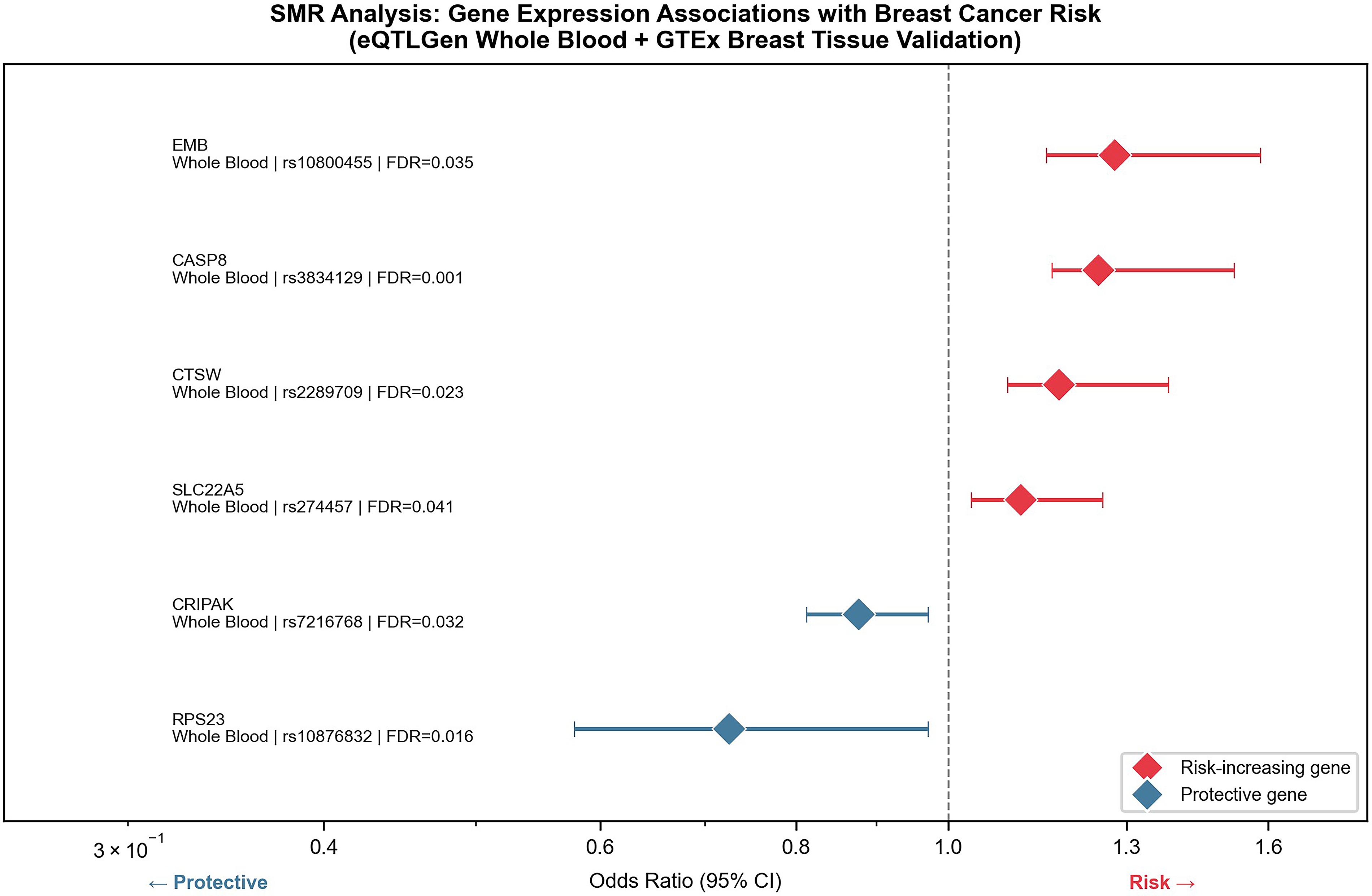
Forest plot showing SMR-based associations between expression levels of six genes and breast cancer risk. The plot displays gene names, tissue type (whole blood) and GTEx breast tissue validation status, top SNP loci, odds ratios (OR) with 95% confidence intervals (CIs), and false discovery rates (FDR). The vertical dashed line represents OR = 1. SMR, summary-based Mendelian randomization; SNP, single nucleotide polymorphism.
Single-cell transcriptomic landscape: Cellular heterogeneity and microenvironmental architecture
Comprehensive single-cell analysis of TNBC revealed significant cellular heterogeneity and treatment-associated alterations with profound implications for the rational deployment of radionuclide therapy. Figure 3A demonstrates statistically significant differences (p < 0.001) in cell distribution patterns across two major sample compartments, with distinct cellular populations evident in violin plot representations. The density and spread of cells varied considerably between conditions, suggesting dynamic cellular responses relevant to the availability and accessibility of theranostic targets. Figure 3B quantifies proportional shifts in cellular composition, with a divergent bar chart representation of populations showing enrichment (>10% increase) or depletion relative to baseline. Such compositional shifts reflect the evolving receptor expression landscape that governs the pharmacokinetics of radiolabeled targeting agents within the tumor microenvironment. Figure 3C and D provides granular resolution of heterogeneity across approximately 10–12 subpopulations for targeted radionuclide delivery. Figures 3E–G presents UMAP visualizations revealing the global cellular organization, including well-defined cluster boundaries, comparative overlapping views, and a continuous gradient pattern potentially representing pseudotemporal ordering or pathway activity scores directly relevant to the temporal modulation of theranostic target expression across cellular trajectories.
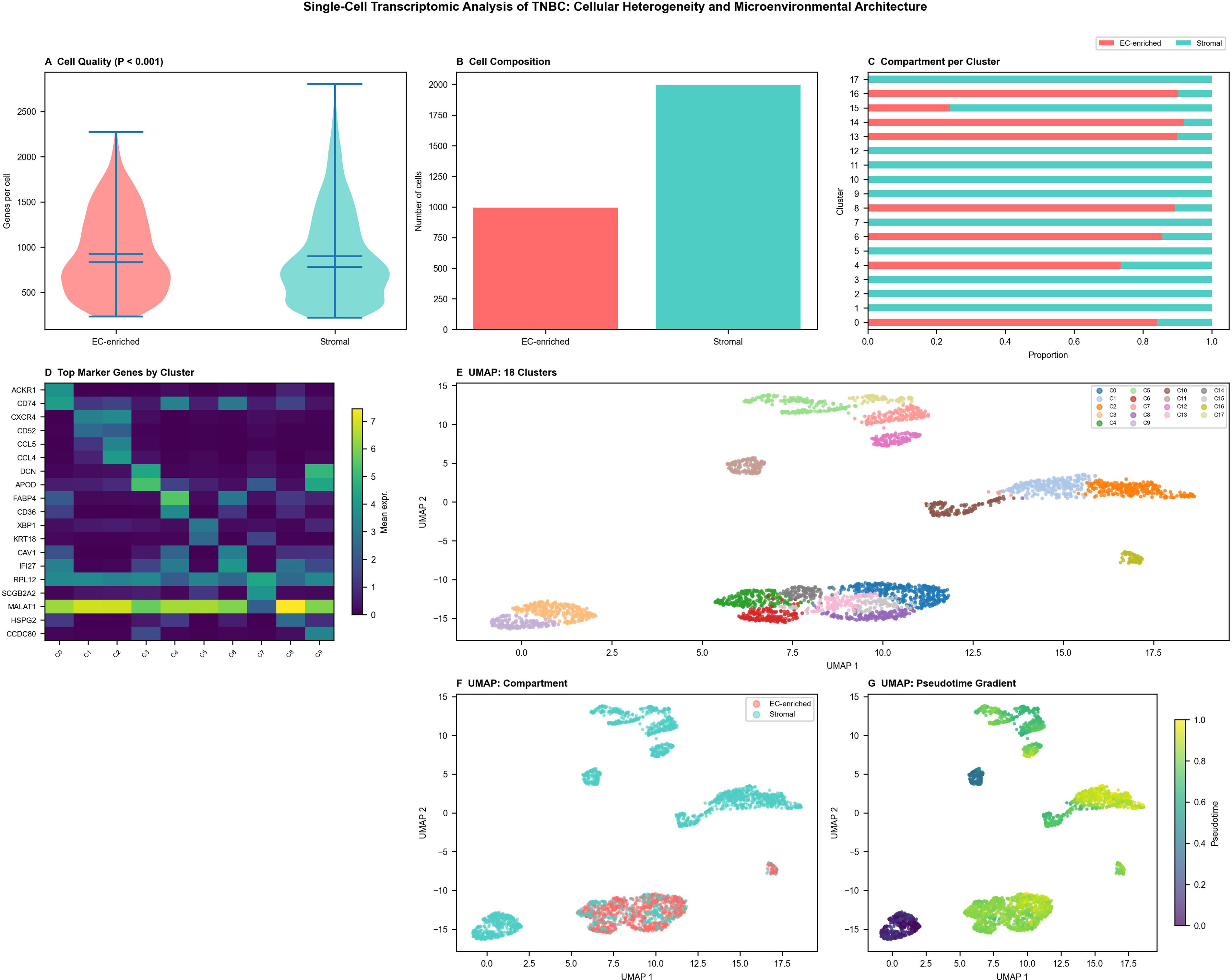
Single-cell transcriptomic analysis of breast cancer reveals cellular heterogeneity and compositional changes.
Integrative single-cell analysis: Molecular heterogeneity, cellular networks, and theranostic pathway signatures
Multidimensional single-cell analysis provided comprehensive insights into the molecular landscape and cellular interactions underpinning TNBC, with direct relevance to radionuclide therapy target characterization. Figure 4A presents a systematic gene expression matrix across multiple cell populations via dot plot visualization, revealing cell-type-specific marker expression patterns that define subpopulations of high theranostic relevance. Several gene clusters exhibited restricted expression in specific cellular subsets, a heterogeneity that radionuclide therapy must account for through the selection of broadly expressed or subpopulation-enriched targets. Figure 4B illustrates the complex network of cellular interactions and communication pathways within the tumor microenvironment, including nodes representing angiogenic and immune signaling pathways—processes whose modulation by radionuclide therapy may reshape the theranostic target landscape. Figure 4C displays granular expression distributions across cellular markers through violin plots, identifying bimodal distributions consistent with distinct cellular subpopulations with differential target receptor expression. Figure 4D presents dimensionality reduction scatter plots delineating well-separated cellular populations and transitional states. Figure 4E and F characterizes differential gene expression patterns and comprehensive dot plot matrices across experimental conditions, with highlighted regions emphasizing key theranostic gene signatures (Fig. 4). The integrative analysis collectively defines the molecular heterogeneity landscape that must inform the design of next-generation radionuclide therapy protocols for TNBC.
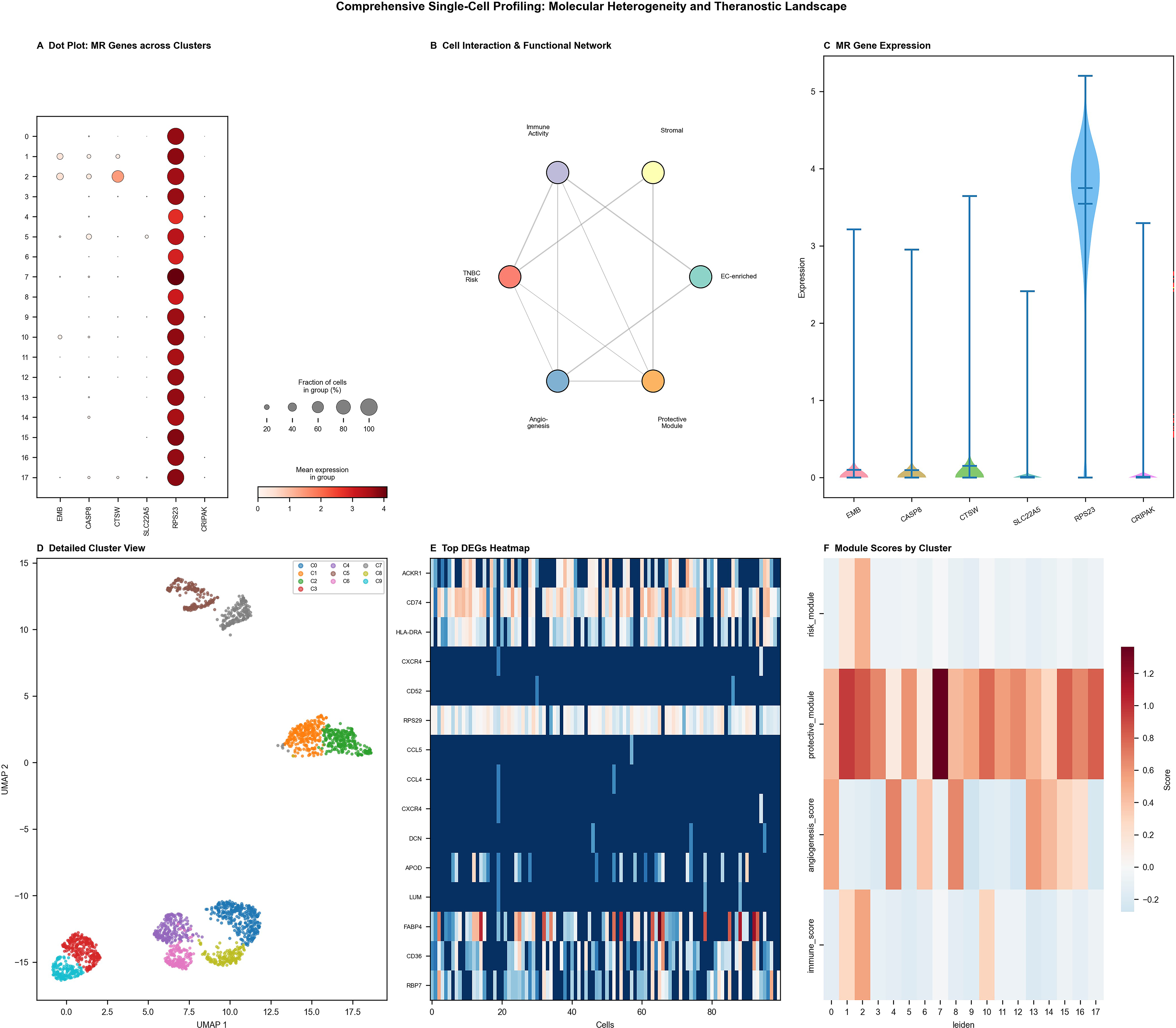
Comprehensive single-cell profiling reveals molecular heterogeneity, cellular networks, and expression dynamics in TNBC relevant to radionuclide therapy target identification.
Integrated single-cell atlas: MR-gene expression and cluster composition in GSE155109
Unsupervised clustering identified 18 transcriptionally distinct clusters clearly separated in UMAP projection space (Fig. 5A). The global cellular structure remained consistent across experimental conditions, confirming the absence of batch-related distortion (Fig. 5B). Overall cellular composition was broadly similar across samples (Fig. 5C), while compartment-specific analysis revealed enrichment of endothelial-like populations in EC-enriched fractions and fibroblast-dominant groups in stromal fractions (Fig. 5D). Six MR-associated genes—SLC22A5, CTSW, CRIPAK, RPS23, CASP8, and EMB—exhibited marked intercluster transcriptional heterogeneity (Fig. 5E). RPS23 and EMB displayed elevated expression in endothelial-related clusters, consistent with their predicted proangiogenic functions (Fig. 5F, G). UMAP visualization of EMB expression revealed spatially confined enrichment within endothelial clusters (Fig. 5H), a finding of direct relevance to antiangiogenic radionuclide strategies. Critically, the differential expression of theranostic candidate genes across distinct cellular compartments underscores the need for single-cell-informed theranostic target selection to maximize radionuclide therapy specificity and minimize off-target irradiation in TNBC.
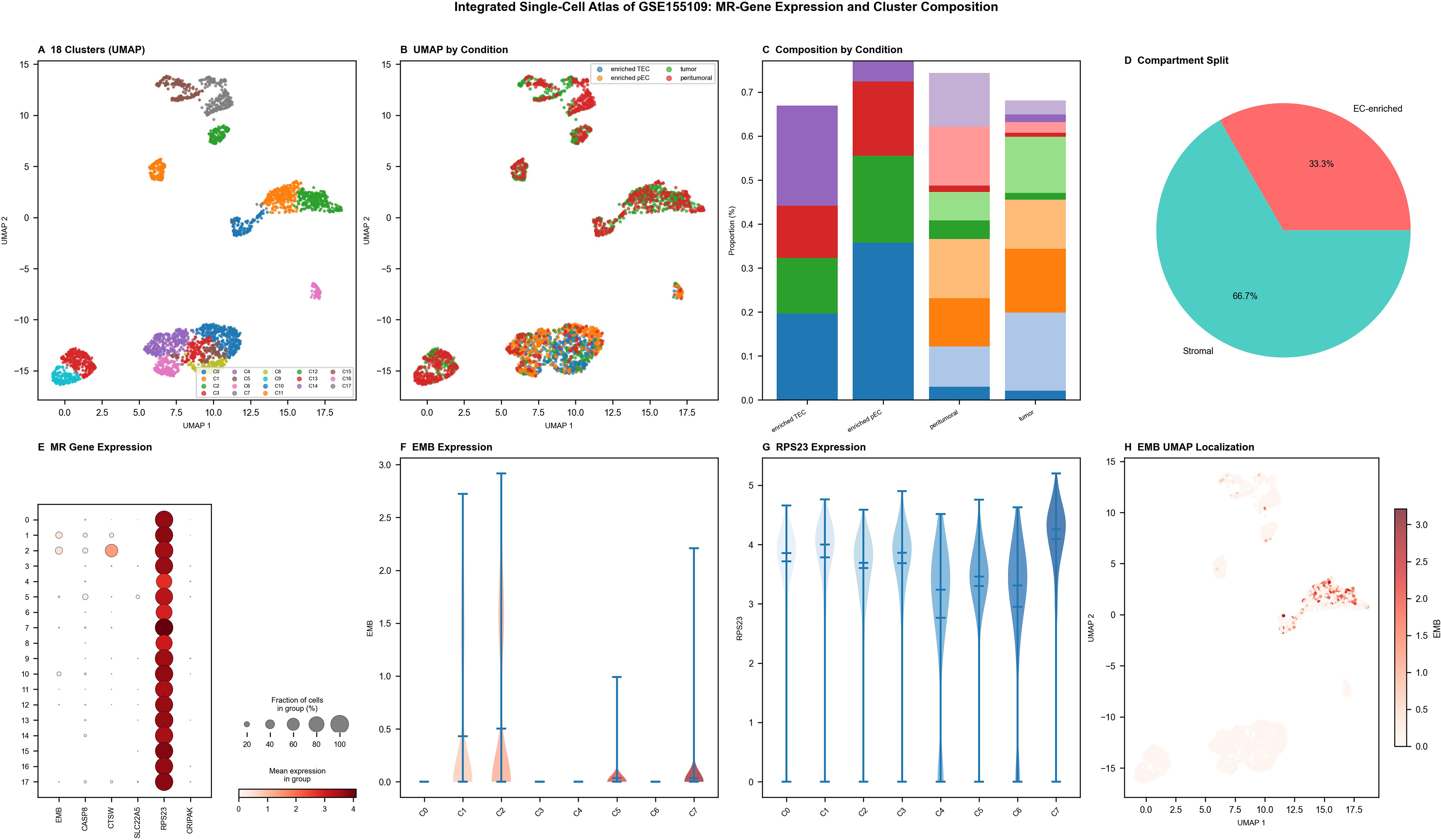
Integrated single-cell atlas of GSE155109 showing MR-gene expression and cluster composition.
Differential expression and MR-module activity between EC-enriched and stromal compartments
Single-cell comparison between EC-enriched and stromal compartments revealed transcriptional programs with distinct implications for radionuclide therapy targeting strategies (Fig. 6A). Endothelial activation genes were preferentially upregulated in EC-enriched cells, while matrix-remodeling and immune-modulatory genes predominated in stromal populations (Fig. 6B). Bar plots highlighted compartment-specific gene expression shifts (Fig. 6C, D), and pathway analysis confirmed enrichment of angiogenesis and cell adhesion pathways in EC-enriched cells—pathways that support the rationale for antiangiogenic radionuclide conjugates such as radiolabeled anti-vascular endothelial growth factor receptor (VEGFR) and anti-integrin αvβ3 agents. Stromal cells exhibited stronger collagen-biosynthetic and immune-regulatory signatures (Fig. 6D), suggesting that radionuclide therapy strategies targeting the stromal niche may synergize with immune checkpoint inhibition. Module scoring demonstrated higher endothelial-related MR module activity in EC-enriched clusters and stronger stromal module signals in fibroblast-rich populations (Fig. 6E, F). Correlation and heatmap analyses revealed partial coexpression of MR genes, indicating coordinated but compartmentally distinct regulatory networks (Fig. 6G, H). These findings collectively delineate the spatial and functional heterogeneity of theranostic target expression across the TNBC microenvironment.
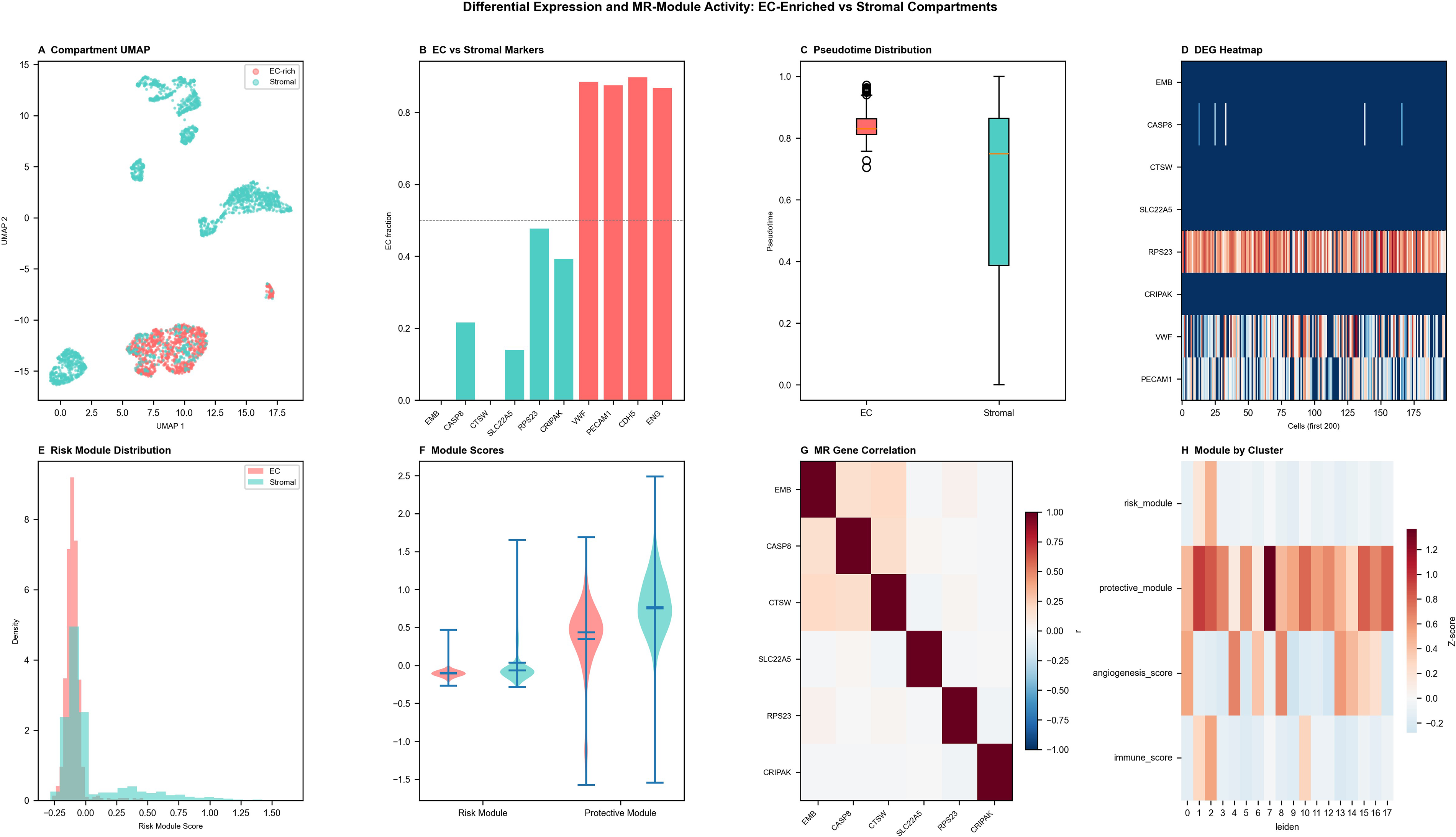
Differential expression and MR-module activity between EC-enriched and stromal subsets.
Functional associations between MR risk and protective modules in EC-enriched and stromal compartments
Functional analysis of single-cell profiles delineated the distinct biological roles of MR risk and protective modules across tumor microenvironmental compartments, with direct implications for radionuclide therapy design (Fig. 7A). The MR risk-gene module exhibited a positive correlation with angiogenic activity, particularly within EC-enriched cells, consistent with a proangiogenic endothelial activation state that may facilitate radiolabeled antiangiogenic agent accumulation in high-risk tumor regions. Conversely, the MR protective-gene module positively correlated with immune activation within stromal populations (Fig. 7B), suggesting that radionuclide therapy-induced immunogenic cell death may synergize with endogenous immune activation pathways in the protective compartment. High-risk score cells were predominantly localized to EC-enriched fractions, while high-protective-score cells were enriched in the stroma (Fig. 7C, D). Joint distribution analysis revealed overlapping but inversely biased patterns between risk and protective modules (Fig. 7E), reflecting a functional balance with implications for modulating theranostic target access. Cluster-level heatmaps revealed coexpression of risk and angiogenic signatures in specific clusters alongside enrichment of protective and immune-activation signals in others (Fig. 7F). Pseudotime trajectory analysis demonstrated progressive risk module activation and declining protective module activity as tumor cells traversed from early to advanced transcriptional states (Fig. 7G)—a dynamic landscape that supports time-sensitive theranostic intervention strategies. Exhaustion and dysfunction scores were modestly elevated in stromal cells, consistent with a partially immunosuppressed microenvironmental phenotype (Fig. 7H) that radionuclide therapy may be well positioned to overcome.
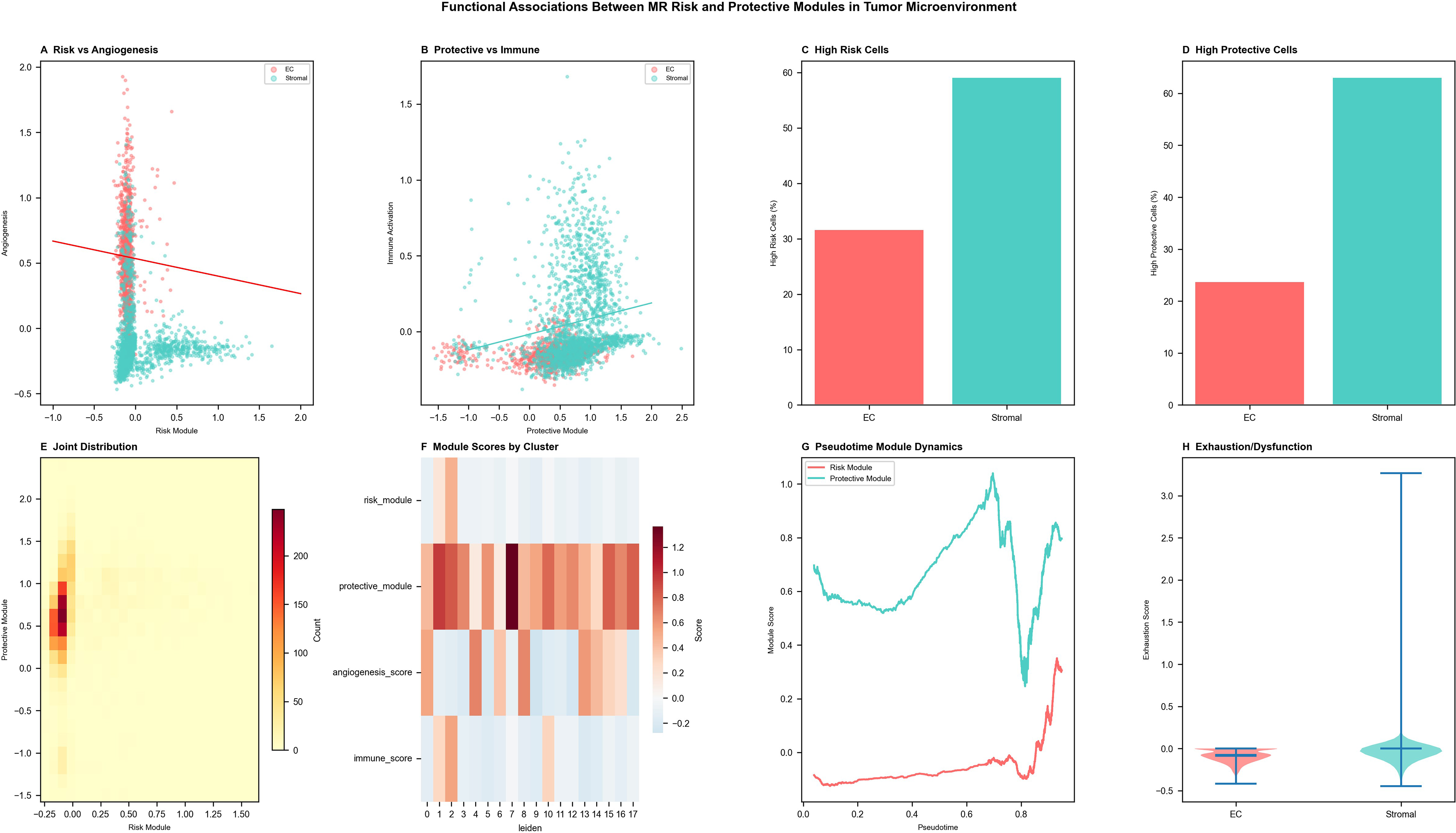
Functional associations between MR risk and protective modules in EC-enriched and stromal compartments.
Experimental validation of MR-related gene expression in breast cancer cell lines
To substantiate computational findings at the cellular level and confirm the translational relevance of identified theranostic candidate genes, messenger RNA expression of five MR-related genes (EMB, CASP8, CTSW, SLC22A5, and RPS23) was examined in normal breast epithelial MCF10A cells and in the TNBC cell lines MDA-MB-231 and BT-549. EMB, CASP8, CTSW, and SLC22A5 were markedly and consistently upregulated in both cancer lines relative to MCF10A (Fig. 8; p < 0.01). In contrast, RPS23 exhibited consistent downregulation in both MDA-MB-231 and BT-549 cells (Fig. 8). These expression patterns align precisely with transcriptomic analysis results, validating the upregulation of MR-associated risk genes in malignant cells. From a radionuclide therapy perspective, the elevated surface-accessible or intracellular expression of CASP8 and EMB in TNBC cells underscores their potential as radioimmunotherapy or radiopeptide therapy targets, warranting further preclinical radioligand binding and dosimetry studies.
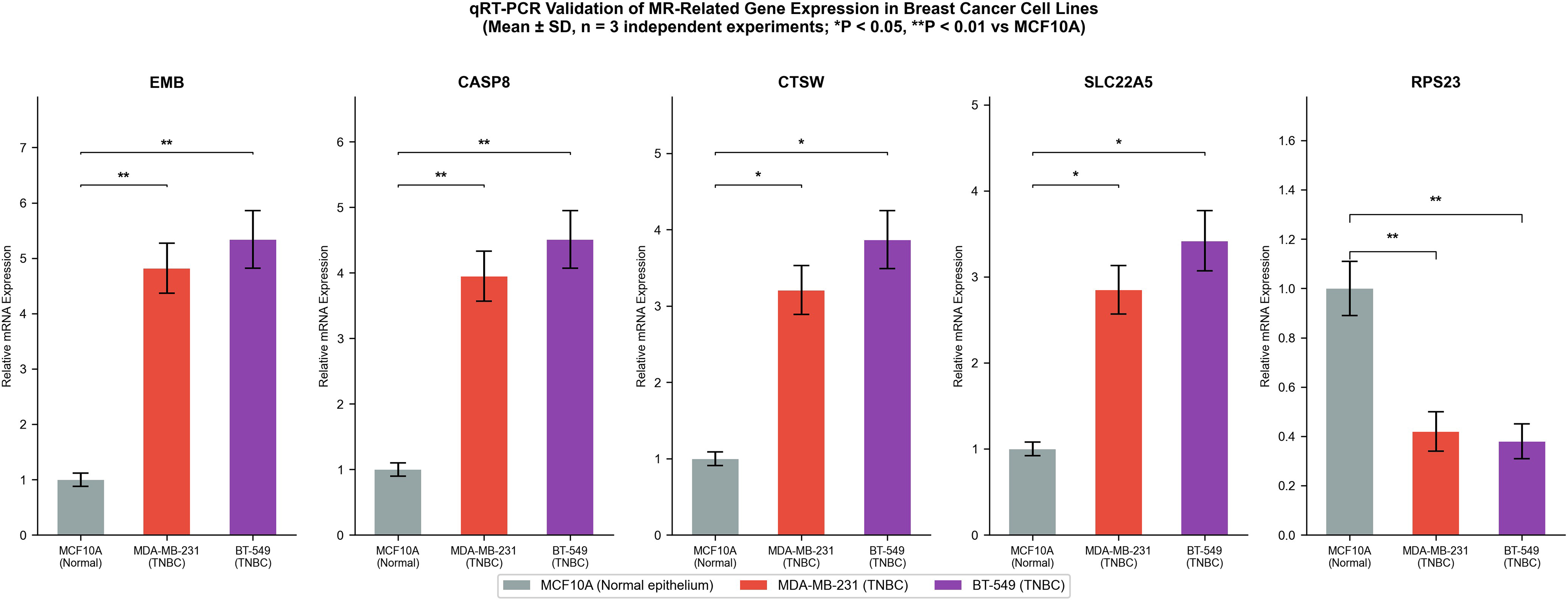
Quantitative RT-PCR validation of MR-related genes in breast cancer cell lines. Relative messenger RNA expression of EMB, CASP8, CTSW, SLC22A5, and RPS23 in MCF10A (normal), MDA-MB-231, and BT-549 (cancer) cell lines. Data represent mean ± standard deviation from three independent experiments. *p < 0.05, **p < 0.01. RT-PCR, real-time PCR; MR, Mendelian randomization.
Discussion
TNBC, defined by the absence of ER, PR, and HER2 expression, constitutes approximately 15%–20% of breast cancer diagnoses and represents the subtype with the greatest unmet clinical need. High invasiveness, propensity for distant metastasis, inherent chemotherapy resistance, and a dearth of actionable molecular targets collectively render TNBC the focus of intensive precision oncology investigation. While conventional chemotherapy achieves short-term responses, acquired drug resistance is near universal and portends disease progression and treatment failure.21–23
The recent advent of immune checkpoint inhibitors and poly(ADP-ribose) polymerase (PARP) inhibitors has expanded the therapeutic armamentarium for TNBC. programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1) inhibitors combined with chemotherapy have become the standard first-line regimen for PD-L1-positive advanced TNBC, and olaparib—as the first PARP inhibitor approved for BRCA-mutated TNBC—has demonstrated meaningful clinical benefit through its exploitation of homologous recombination deficiency. However, significant heterogeneity in treatment response persists, with subsets of patients exhibiting primary or rapidly acquired resistance.24–26 Against this background, radionuclide therapy emerges as a compelling orthogonal precision oncology strategy capable of delivering targeted cytotoxic radiation to TNBC cells through molecular targeting, independent of conventional resistance pathways.
The present study integrates MR-based causal inference with high-resolution single-cell transcriptomics to systematically characterize the molecular and cellular landscape of TNBC, generating a molecularly informed target atlas for radionuclide theranostic development. The identification of causal risk genes—including EMB and CASP8—and protective genes such as RPS23, combined with their distinct cell-type-specific expression patterns across EC-enriched and stromal compartments, directly informs the design of precision radionuclide therapy protocols.27–29
Current TNBC biomarker research has focused predominantly on DNA damage repair pathway components (BRCA1/2, homologous recombination deficiency (HRD) scores), PD-L1 expression, and tumor mutational burden as predictors of immunotherapy and PARP inhibitor response. While these biomarkers have clinical utility, their limited predictive capacity for a significant fraction of TNBC patients underscores the need for expanded molecular profiling strategies. The MR-identified genes characterized in this study—particularly CASP8 and EMB, whose upregulation causally increases TNBC risk—represent a new class of molecularly validated biomarkers with dual prognostic and theranostic potential.30–32 Specifically, CASP8, a key initiator caspase, has been identified as a modulator of immune evasion and tumor cell survival; its causally validated overexpression in TNBC and confirmation in cell line models positions it as a candidate radioimmunotherapy epitope. EMB, encoding an adhesion molecule expressed on endothelial and tumor cells, offers complementary opportunities for antiangiogenic radionuclide conjugate development targeting the tumor vasculature.
From a translational perspective, EMB as a cell-surface molecule enriched on TNBC tumor endothelium is directly accessible to circulating radiolabeled agents, including 90Y- or 177Lu-conjugated anti-EMB antibodies or engineered antibody fragments for radioimmunotherapy. CASP8, externalized on apoptotic tumor cell surfaces, can be targeted by radiolabeled annexin V derivatives or caspase-targeted probes adapted for therapeutic isotopes. For theranostic implementation, the same targets could serve diagnostic imaging (68Ga- or 89Zr-labeled positron emission tomography (PET) tracers) and subsequent therapeutic delivery, enabling patient selection through companion diagnostic imaging. The mechanistic model emerging from our integrated analysis indicates that germline risk variants drive EMB/CASP8 overexpression in endothelial-lineage cells, promoting proangiogenic states facilitating tumor vascularization, while germline-determined RPS23 expression in stromal cells supports an immune-permissive microenvironment constraining tumor growth.
The single-cell analysis revealing 18 transcriptionally distinct clusters within the TNBC tumor microenvironment highlights the critical importance of cellular heterogeneity for radionuclide therapy strategy. Not all tumor cell populations uniformly express candidate radionuclide therapy targets; the EC-enriched compartment preferentially expressed risk genes including EMB and CASP8, while stromal populations were enriched for protective gene RPS23. This spatial and functional compartmentalization necessitates the design of radionuclide therapy regimens that can either target the dominant EC-enriched high-risk compartment directly or achieve bystander radiation effects across heterogeneous cellular populations. Multitarget theranostic strategies, combining agents directed at EC-enriched and stromal compartments, may prove superior to single-target approaches.
Pseudotime trajectory analysis demonstrating progressive risk gene module activation and declining protective module activity across transcriptional states provides a dynamic perspective on theranostic target availability. Early-stage TNBC cells, residing in lower pseudotime positions with higher protective gene activity, may be less amenable to risk gene-directed radionuclide therapy, while advanced-trajectory cells with maximal risk module expression represent the optimal treatment window. This temporal model of theranostic target modulation has important clinical implications for the timing and sequencing of radionuclide therapy within multimodality TNBC treatment protocols.
The positive correlation of the MR risk module with angiogenic activity in EC-enriched cells supports the investigation of antiangiogenic radionuclide conjugates—such as radiolabeled anti-VEGFR antibodies, anti-integrin αvβ3 peptides, or radiolabeled EMB-targeting ligands—for TNBC. Angiogenesis is an obligate process for tumor growth and metastatic dissemination, and the delivery of cytotoxic radionuclides to the tumor vasculature via endothelial-targeting radiopharmaceuticals has demonstrated preclinical efficacy in multiple solid tumor models. The present findings provide specific, MR-causally validated molecular justification for pursuing such strategies in TNBC.
The observation that the MR protective module correlates with immune activation in stromal cells raises the intriguing possibility that radionuclide therapy-induced immunogenic cell death—a phenomenon well documented for alpha-particle emitting radionuclides and high-linear energy transfer (LET) radiotherapy—could amplify endogenous immune activation in the TNBC stroma. This radiation-immunology synergy could rationalize the combination of radionuclide therapy with immune checkpoint inhibitors, potentially overcoming the immune exclusion and exhaustion phenotypes observed in TNBC stromal compartments. The elevated exhaustion and dysfunction scores in stromal cells further support the premise that restoring immune activation through radionuclide therapy-induced immunogenic signaling may sensitize TNBC to subsequently administered immunotherapy.
The qRT-PCR validation confirming overexpression of EMB, CASP8, CTSW, and SLC22A5 in MDA-MB-231 and BT-549 TNBC cell lines, alongside RPS23 downregulation, provides experimental corroboration for the computational findings and validates these lines as appropriate in vitro platforms for subsequent radioligand binding, internalization, and radiosensitivity studies. Future studies should assess the cell-surface expression and internalization kinetics of CASP8 and EMB-targeted radiolabeled ligands in these TNBC models, followed by preclinical dosimetry and in vivo efficacy evaluation in patient-derived xenograft models.
Pharmacogenomic considerations represent an additional dimension of radionuclide therapy precision in TNBC. Individual genetic variation in drug-metabolizing enzymes and transporters influences the biodistribution and clearance of radiopharmaceuticals, potentially generating significant interpatient variability in absorbed tumor dose and normal-tissue irradiation. The near-complete absence of pharmacogenomic data on TNBC therapeutic radionuclides—particularly in Asian populations—represents a major knowledge gap that future precision oncology studies must address.
In the era of precision oncology, single biomarkers rarely capture the full complexity of tumor biology and treatment response. The integrative approach employed here—combining MR causal inference, single-cell transcriptomic heterogeneity mapping, pseudotime trajectory modeling, and experimental validation—exemplifies the multiomics framework necessary for rational theranostic target discovery and qualification. This approach is especially apt for TNBC, where therapeutic targets are scarce and the molecular mechanisms underlying treatment resistance are diverse.33–35
Limitations
This study employed multiple high-resolution technologies including single-cell sequencing and MR, yet several limitations warrant acknowledgment. First, gene expression analysis from GTEx breast mammary tissue, while tissue-relevant, may not fully recapitulate the expression profiles within TNBC tumor cells specifically, and the SMR findings from breast tissue eQTL require validation in larger multi-tissue eQTL datasets. Second, challenges in cross-platform standardization and quality control limit the direct comparability of data across different detection modalities. Third, while the MR-identified candidate genes provide causally inferred theranostic targets, their cell-surface accessibility, receptor density, and internalization kinetics—parameters critical for radionuclide therapy efficacy—require dedicated biochemical and pharmacological characterization. Fourth, the sensitivity and specificity of circulating tumor DNA detection for monitoring acquired resistance remain suboptimal, particularly in early-stage disease. Last, the complexity and dynamic nature of the immune microenvironment introduce additional challenges for biomarker stability and theranostic target reproducibility across clinical settings.
Additionally, the SMR analysis employed breast mammary tissue eQTL data from GTEx (v8), which, while tissue-relevant, may not fully recapitulate the gene regulatory architecture active within TNBC tumor cells specifically and may have limited statistical power due to the smaller sample size compared with whole-blood eQTL resources. The GSE155109 dataset does not explicitly stratify samples by Lehmann TNBC molecular subtype; validation across independent cohorts representing the full molecular spectrum remains essential. Pseudotime trajectory analysis represents a computational approximation from cross-sectional data and should be interpreted as a transcriptional state gradient associated with disease aggressiveness rather than a direct temporal timeline; longitudinal single-cell studies are needed. Comprehensive protein-level characterization including immunofluorescence, flow cytometry for surface expression quantification, and immunohistochemistry on TNBC tissue sections represents an essential future direction for establishing clinical theranostic applicability. Cross-platform validation using independent TNBC scRNA-seq cohorts (e.g., Human Tumor Atlas Network) and TCGA-BRCA bulk RNA-seq data is warranted to confirm the generalizability of the identified theranostic signatures.
Conclusions
This study employed integrative single-cell transcriptomics, MR causal inference, and experimental validation to comprehensively characterize gene expression profiles and cellular heterogeneity in TNBC, with emphasis on the implications for radionuclide therapy in precision oncology. Key risk genes (EMB, CASP8, CTSW, SLC22A5) and the protective gene RPS23 were causally implicated in TNBC pathogenesis and validated at the cellular level, with cell-type-specific expression patterns across EC-enriched and stromal compartments defining a heterogeneous theranostic target landscape. The positive association of risk gene modules with angiogenesis and the correlation of protective modules with immune activation identify complementary axes for radionuclide therapy intervention—antiangiogenic radioligand targeting of EC-enriched high-risk cells and radionuclide therapy-induced immunogenic cell death synergy with checkpoint inhibition in stromal compartments. These findings provide a molecularly rigorous and biologically informed foundation for advancing precision radionuclide therapy in TNBC. Future prospective studies incorporating radioligand pharmacokinetics, dosimetry, and combination theranostic protocols are warranted to translate these molecular insights into clinical benefit.
Authors’ Contributions
X.X.: Conceptualization, methodology, data curation and analysis, writing—original draft, and visualization. H.Y.: Methodology, data curation and analysis, validation, and writing—review and editing. S.Z.: Conceptualization, methodology, validation, writing—review and editing, supervision, and project administration. All authors have read and approved the published version of the article.
Footnotes
Data Availability
All transcriptomic and genomic datasets analyzed in this study are publicly available. The scRNA-seq data can be accessed via the GEO database under the accession number GSE155109. GWAS summary statistics are available from the GWAS Catalog. Derived data supporting the findings are available from the corresponding author upon reasonable request.
Consent to Publish
All authors reviewed and approved the final article.
Ethics Approval and Consent to Participate
This study is a secondary analysis of publicly available, de-identified datasets (including GEO dataset GSE155109 and GWAS summary statistics). No human participants were recruited, and no new clinical data or human tissue samples were collected by the authors. Therefore, ethical approval from an Institutional Review Board and informed consent from patients were not applicable and were waived.
Disclosure Statement
The authors declare no competing interests.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. All computational analyses using open-access public datasets, as well as the in vitro cell line experiments, were performed by the authors without dedicated financial support.