
Abstract
Introduction
In the past, ADHD was thought to be confined to children and teens. More recent research has revealed that symptoms can persist during adulthood for up to two thirds of patients (Biederman, Petty, Evans, Small, & Faraone, 2010; Kooij et al., 2010). Approved medications for adult ADHD are the sustained-release stimulants (methylphenidate or amphetamines) and the non-stimulant atomoxetine. Although the stimulants methylphenidate and amphetamine have been shown to be safe and effective for the treatment of adult ADHD (Seixas, Weiss, & Muller, 2012), their utility is limited by concerns over potential abuse, misuse, or diversion (Wilens et al., 2008). Atomoxetine, though effective in adult ADHD (Adler et al., 2009), requires titration and can take several weeks to exert its clinical effect after each dosage adjustment (Michelson et al., 2003). In addition, it carries warnings regarding suicidal ideation and liver toxicity (Capuano et al., 2014).
The limitations of the approved medications for adult ADHD highlight the need for an effective and well-tolerated non-stimulant medication. One such medication under study is the synthetic catecholamine droxidopa, which is decarboxylated by DOPA decarboxylase (DDC) and is converted directly to norepinephrine (NE). Droxidopa can cross the blood–brain barrier (BBB), resulting in increased levels of NE in the central nervous system (CNS) as well as peripherally (Goldstein, 2006). Droxidopa (NORTHERA, Lundbeck LLC, Deerfield, IL, USA) was recently approved by the U.S. Food and Drug Administration (FDA) for the treatment of symptomatic neurogenic hypotension caused by primary autonomic failure (Parkinson’s disease, multiple system atrophy, and pure autonomic failure).
The primary objective of our study was to determine the effect of droxidopa therapy, alone and in combination with carbidopa, on adult ADHD symptoms over the course of a 6-week, open-label titration period followed by a 2-week, double-blind, placebo-controlled period. Carbidopa is a DOPA decarboxylase inhibitor (DDCI) that cannot cross the BBB. Inhibition of peripheral DDC through carbidopa co-treatment may enable increased central levels of NE (Kaufmann, 2006), potentially increasing CNS response while limiting the potential for adverse events (AEs) in the periphery. The combination therapy was expected to have an acceptable safety profile, as concomitant use of a DDCI with droxidopa in patients with Parkinson’s disease had no significant impact on safety of the study treatment in a randomized, placebo-controlled, double-blind, Phase II trial conducted at 30 European centers (Mathias, 2008).
Method
Patients
Patients were enrolled from a single study site, Veterans Affairs New York Harbor Healthcare System, New York University Langone Medical Center, New York, NY, USA, between January 2010 and May 2011. Eligible patients were men and women aged 18 to 55 years with ADHD and no clinically significant abnormalities as determined by medical history, physical examination, electrocardiogram (ECG), and clinical laboratory testing. The diagnosis of ADHD was confirmed using the Adult ADHD Clinician Diagnostic Scale (ACDS, v 1.2; Adler & Cohen, 2004); other Axis I psychiatric disorders were evaluated using the Structured Clinical Interview for DSM-IV-TR Axis I Disorders Research Version, Patient Edition (SCID-I/P; First, Spitzer, Gibbon, & Williams, 2002). Allowed concomitant Axis I diagnoses included social anxiety disorder or dysthymia not requiring treatment. The exclusion criteria included history of bipolar or psychotic disorders; uncontrolled comorbid major depressive disorder, anxiety disorder, or dysthymia; regular use of any nonhypnotic psychotropic agents; and significant cardiac, hepatic, renal, or other systemic illness.
Study Design
The conduct of this pilot trial was approved by the Subcommittee for Human Subjects at the VA New York Harbor Healthcare System. The study was conducted in accordance with the Declaration of Helsinki and International Conferences on Harmonisation Good Clinical Practices Guidelines and was in compliance with applicable regulatory guidelines and regulations. All patients gave written informed consent to participate. AEs were regularly monitored; however, a Data and Safety Monitoring Board (DSMB) was not employed as this was a single-site study that did not evaluate mortality or major morbidity as outcomes (FDA, 2006).
Droxidopa (200 mg capsules), carbidopa (25 mg capsules), and matching droxidopa placebo and carbidopa placebo capsules were provided by Lundbeck NA Ltd., Charlotte, NC, USA. Carry-over effects from previously taken psychotropic medications were minimized by including a 4-week washout period (1 week for psychostimulants) prior to enrollment, with the exception of “as needed” use of benzodiazepines and hypnotics, which were allowed during the study.
This was a single-center, 12-week, two-period study conducted under IND#103,627 from the FDA (ClinicalTrials.gov: NCT00983814). During the first period, patients underwent 6 weeks of open-label treatment (Figure 1) following a 1-week screening. In the first 3 weeks, droxidopa monotherapy was titrated from 200 mg 3 times per day (TID) up to a maximum of 600 mg TID, depending on clinical response and tolerability. This was followed by 3 weeks of fixed-dose treatment with droxidopa (at the previously determined tolerable level) plus concomitant carbidopa, which was titrated from 25 to 50 mg TID. In the second study period, patients completing open-label treatment were then randomized by code envelopes in a 1:1 ratio either to continued combination treatment or to matching placebo for 2 weeks under double-blind conditions. Those randomized to treatment continued to receive the same fixed-dose droxidopa/carbidopa regimen that they had been receiving at the end of the open-label treatment period.
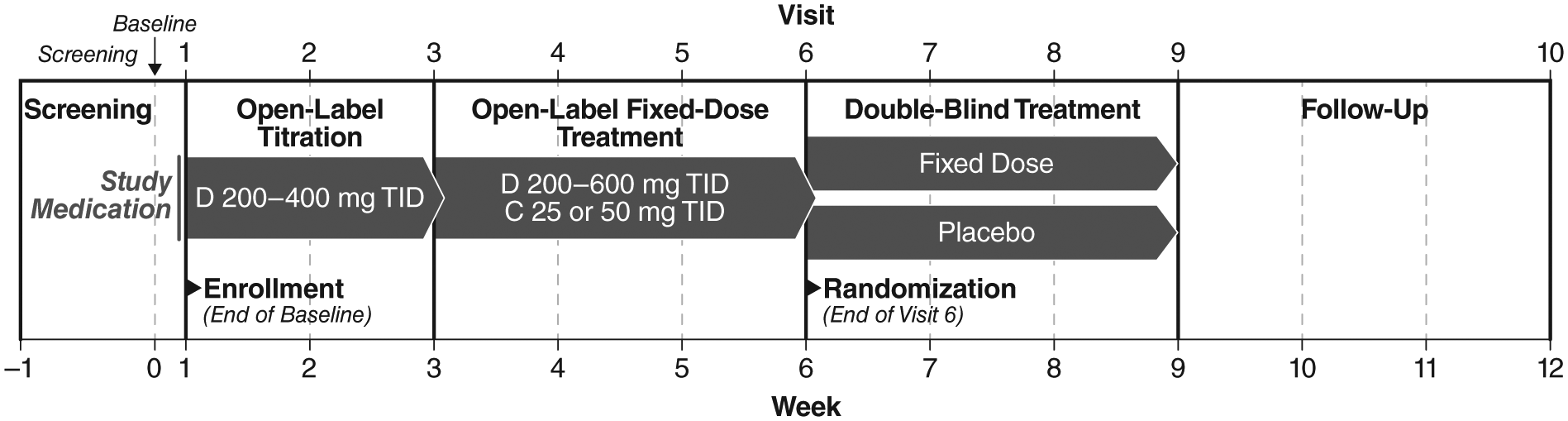
Two-period study design.
Patients completed a total of 12 scheduled visits during this two-period study: screening, baseline, 8 weekly treatment visits (Week 1 through Week 8), and two follow-up visits (Visit 9 at Week 9 and Visit 10 at Week 12).
Efficacy Endpoints and Assessments
Efficacy was evaluated at each study visit. The primary efficacy outcome measure was change from baseline in the mean total score on the Adult ADHD Investigator Symptom Report Scale (AISRS; Spencer et al., 2010). The AISRS is an 18-item, validated, semi-structured, investigator-administered instrument for the assessment of ADHD symptoms. The severity of each of the items is rated on a 4-point scale (none, mild, moderate, severe). The AISRS has been used in numerous adult ADHD treatment trials (Adler et al., 2011; Adler et al., 2009).
The secondary efficacy endpoints included changes from baseline in the mean Inattentive Subset and Hyperactive Impulsive Subset scores on AISRS as well as changes from baseline in self-reported ADHD symptoms, as assessed by mean Total score, mean Inattentive Subset score, and mean Hyperactive Impulsive Subset score on the Adult ADHD Self-Report Scale (ASRS, v 1.1; Adler et al., 2006). The ASRS is a validated, self-administered instrument for the assessment of ADHD symptoms. It comprised 18 items, which are rated by the patient on a 5-point scale (0 = never, 1 = rarely, 2 = sometimes, 3 = often, 4 = very often). The secondary efficacy endpoints also included change from baseline in the global severity of ADHD as assessed by the Clinician Global Impression (CGI) scale (Guy, 1976). The CGI is a 7-point scale (1 = normal, 2 = borderline, 3 = mild, 4 = moderate, 5 = marked, 6 = severe, 7 = extremely severe).
Safety Assessments
AEs were assessed at each study visit. An AE was defined as any adverse change from the patient’s baseline (pre-treatment) condition that occurred during the study after treatment had started. A serious adverse event (SAE) was defined as death, initial, or prolonged inpatient hospitalization, a life-threatening experience, persistent or significant disability/incapacity, or other medically important event. Other safety parameters included vital signs, ECG, routine clinical laboratory parameters (hematology, blood chemistry, and urinalysis), and serum and urine Troponin I and catecholamine levels. Concomitant medications were monitored at screening/baseline and at each study visit. Suicidal ideation and behavior were monitored carefully, as in all ADHD clinical trials. The Columbia Suicide Severity Rating Scale (Posner et al., 2011) was administered at each study visit. Safety follow-up continued for 30 days following the last study-drug intake.
Statistical Analysis
This small, pilot trial was considered exploratory and hypothesis generating in nature. The goal was to detect signals of efficacy for droxidopa treatment followed by a combination regimen with carbidopa. Twenty patients were expected to complete this study. This sample size was calculated to be sufficient to detect a signal of treatment based on the expected effects of droxidopa from other neuropsychiatric disorders and upon sample sizes from other proof-of-concept trials in adult ADHD (Martin, Corcoran, Zhang, & Katic, 2014; Surman et al., 2013). In a one-way ANOVA, a sample size of 20 (10 per treatment group) gives 80% power to detect an effect size of 0.6625 using an F test with 0.05 significance and standard deviation (SD) of 0.5.
The safety population included all participants enrolled in the study who received at least one dose of study-drug. The primary population for the efficacy analyses was the intent-to-treat (ITT) population (all enrolled patients). However, efficacy analyses at each time point were conducted only on the subset of the ITT population still receiving study medication at that time point. Missing values for questions in the questionnaires were imputed using the “last observation carried forward” method when needed for patients still receiving medication at a given time point. Only post-baseline question responses were used for imputation. This modified-ITT analysis strategy was chosen because the study medication regimen for any individual patient was not constant from one week to the next. Therefore, imputing of missing data for discontinued patients may have led to a distortion of the data, as it would not have accounted for the variation that may have been observed with a change in medication dosage. This effect could have potentially been magnified due to the high discontinuation rate (>20%) in this small, exploratory study. This approach was seen as reasonable for an exploratory study, given that the primary objective was to determine if there was sufficient evidence of a signal of effectiveness for droxidopa in the treatment of adult ADHD symptoms to warrant further, more comprehensive investigations.
All statistical analyses were conducted with the SAS software package (v 9.1.3; Cary, NC, USA) or later. AISRS scores were summarized with descriptive statistics on the raw score and change from baseline by study visit. Time-points associated with the open-label titration and fixed-dose periods (baseline through Week 6) were assessed by the Wilcoxon-signed rank test for change from baseline under the null hypothesis of no change. Time-points in the double-blind and follow-up periods (Weeks 7-12) were assessed by time point for differences between the placebo and active treatment groups using a t test on change from baseline values between the treatment groups.
Results
Study Population
Figure 2 shows the disposition of patients in this study. Thirty-nine patients were screened and 20 patients enrolled over an approximate 1-year period. Thirteen of the 20 enrolled patients completed the 6-week, open-label period. Of these, 11 were randomized to either active treatment with droxidopa/carbidopa (n = 6) or to placebo (n = 5) in the double-blind period. Only 10 of the 11 randomized patients completed the double-blind period, because 1 patient in the placebo group withdrew consent (see “Discontinuations” section). All patients were followed for 30 days after last study-drug administration.
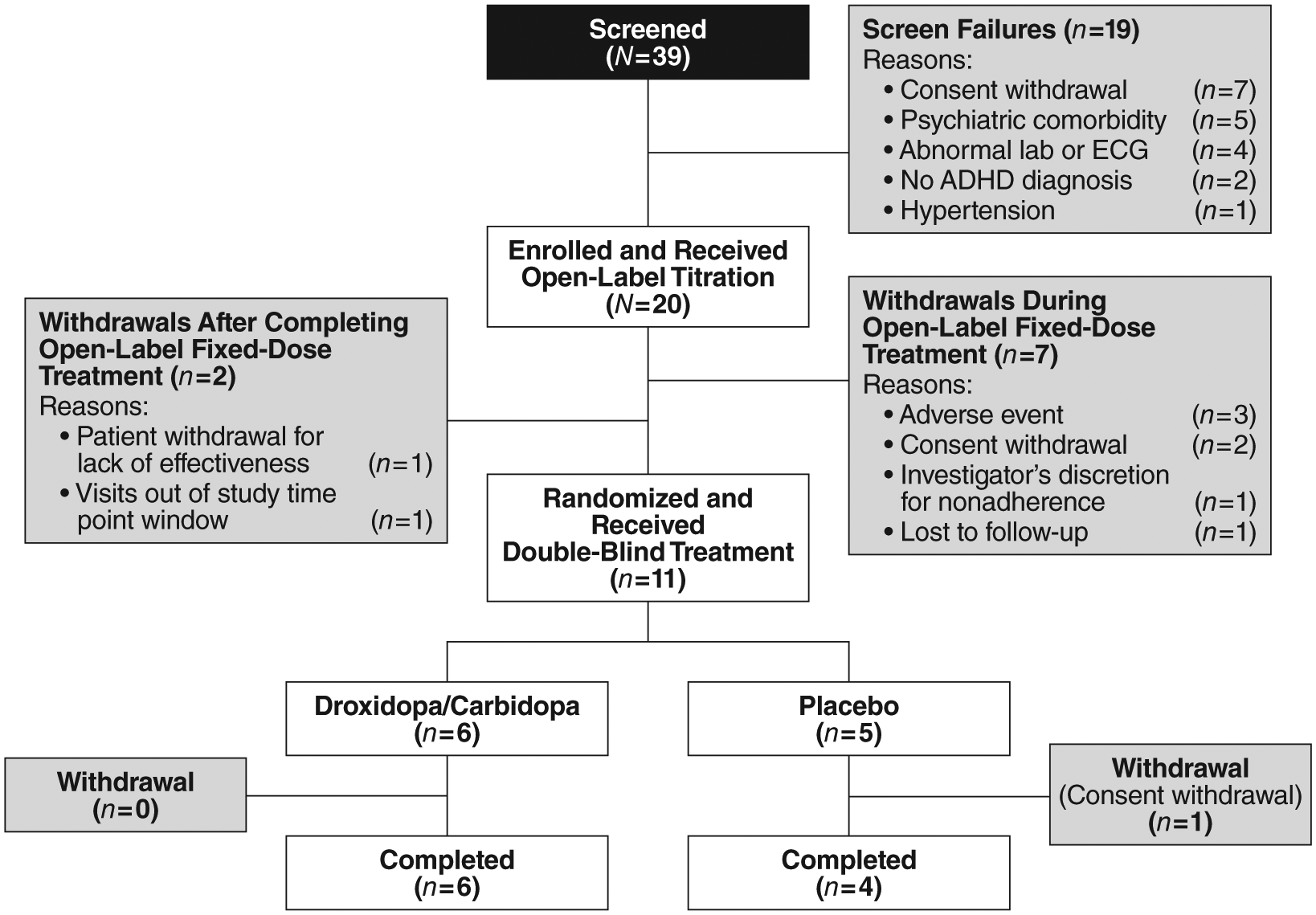
Patient flow and nonadherence.
Baseline characteristics are summarized in Table 1. Eight patients (40%) reported a history of psychiatric disorders in addition to ADHD. These were depression (n = 7 patients), adjustment disorder (n = 2), anxiety (n = 1), and social phobia (n = 1). However, these patients were not receiving treatment for these conditions and only one patient was taking psychoactive medication at baseline (trazodone for chronic insomnia and not depression).
Baseline Patient Characteristics.
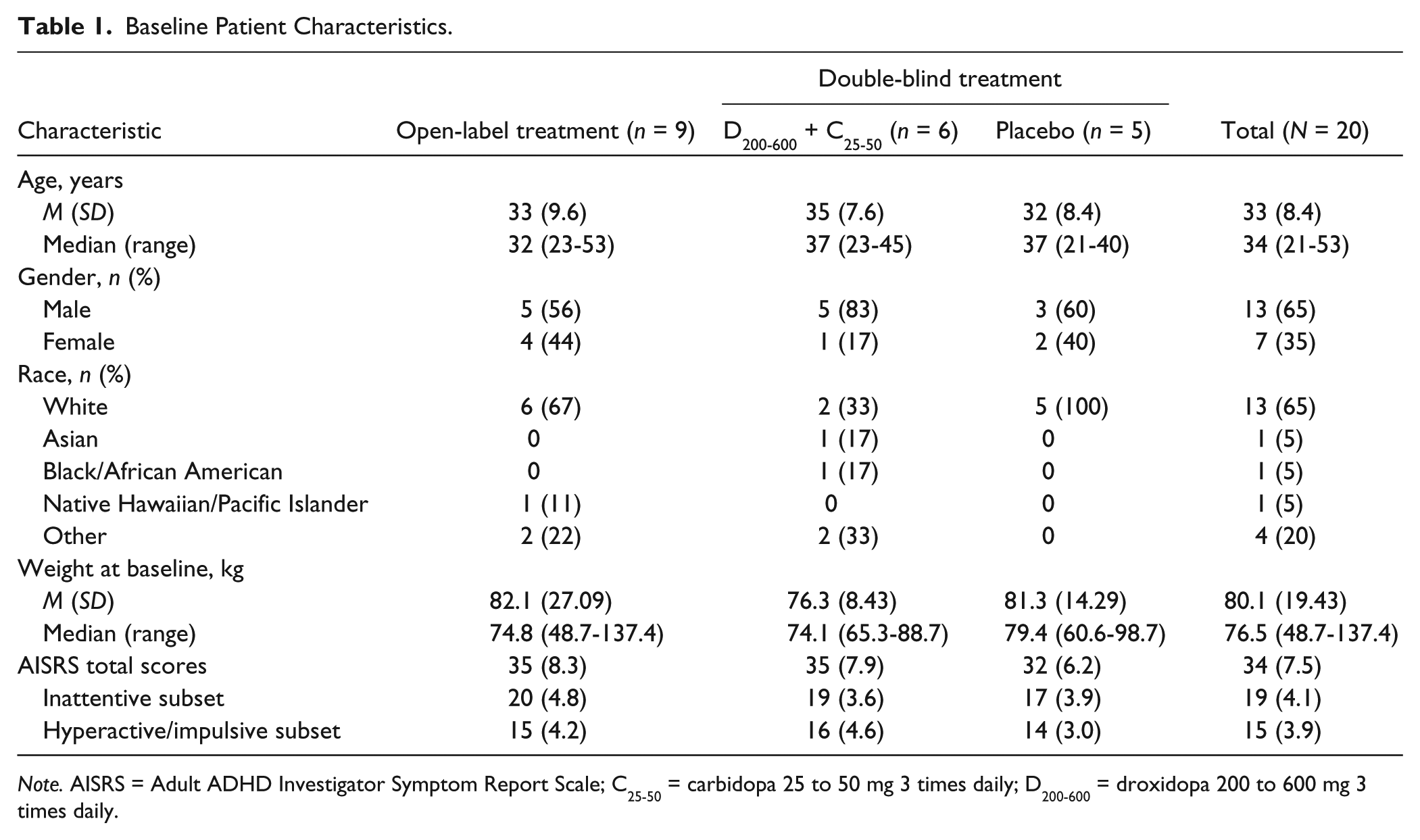
Note. AISRS = Adult ADHD Investigator Symptom Report Scale; C25-50 = carbidopa 25 to 50 mg 3 times daily; D200-600 = droxidopa 200 to 600 mg 3 times daily.
Discontinuations
The majority of the withdrawals (9 out of 20 patients, 45%) occurred during the open-label period (Figure 2). Reasons for discontinuation during the open-label period were consent withdrawal (n = 2), AE (n = 3), visits out of study time window (n = 1), patient-instigated withdrawal due to perceived lack of effectiveness (n = 1), investigator-instigated withdrawal due to nonadherence (n = 1), and lost to follow-up (n = 1). Only 1 patient discontinued in the double-blind period; this patient withdrew consent.
Three patients (15%) discontinued during the open-label period due to AEs. The first patient, a 24-year-old White female, experienced arrhythmia with ECG changes, which was not observed at baseline, beginning the first day of open-label droxidopa monotherapy. The event was assessed as mild in severity and possibly study-drug related. After discontinuation, her symptoms resolved in 23 days. The second patient, a 53-year-old White male, experienced suicidal ideation that was not observed at baseline “without intent or plan” and violence-related ideation expressed as “a desire to punch a wall,” with no action taken after 6 weeks of open-label droxidopa treatment (3 weeks of droxidopa monotherapy, followed by 3 weeks of combined droxidopa/carbidopa treatment). The events were assessed as severe and probably study-drug related. There was no suicide attempt and violent actions did not occur, and symptoms resolved within 30 min of onset. The third patient, a 26-year-old female (race noted as Chinese/White), had an ECG T-wave inversion detected on Day 16 of open-label droxidopa titration. The event was considered moderate in severity and probably study-drug related. The T-wave inversion resolved within 7 days after onset. The patient did not experience any cardiac-related symptoms.
Primary Efficacy Endpoint: Total AISRS Score
The baseline mean ± SD total AISRS score was 34 ± 7.5. As seen in Figure 3, a fairly rapid treatment effect was detected with droxidopa during open-label titration, as shown by a decrease to a mean total AISRS score of 28 ± 11.0 at Week 1 (mean change from baseline: −6 ± 5.9; p < .0001) and 19 ± 12.3 at Week 3 (mean change from baseline: −16 ± 7.7; p < .0001). Scores remained stable, but did not further improve, with the addition of open-label carbidopa (25 or 50 mg). Overall, for the open-label period, the mean total AISRS score at Week 6 was 17 ± 10.5 (mean change from baseline: −18 ± 6.3; p = .0002). A 30% or greater reduction in total AISRS score from baseline was achieved by 13 out of 15 patients (86.7%) at Week 3 and 11 out of 13 patients (84.6%) at Week 6.
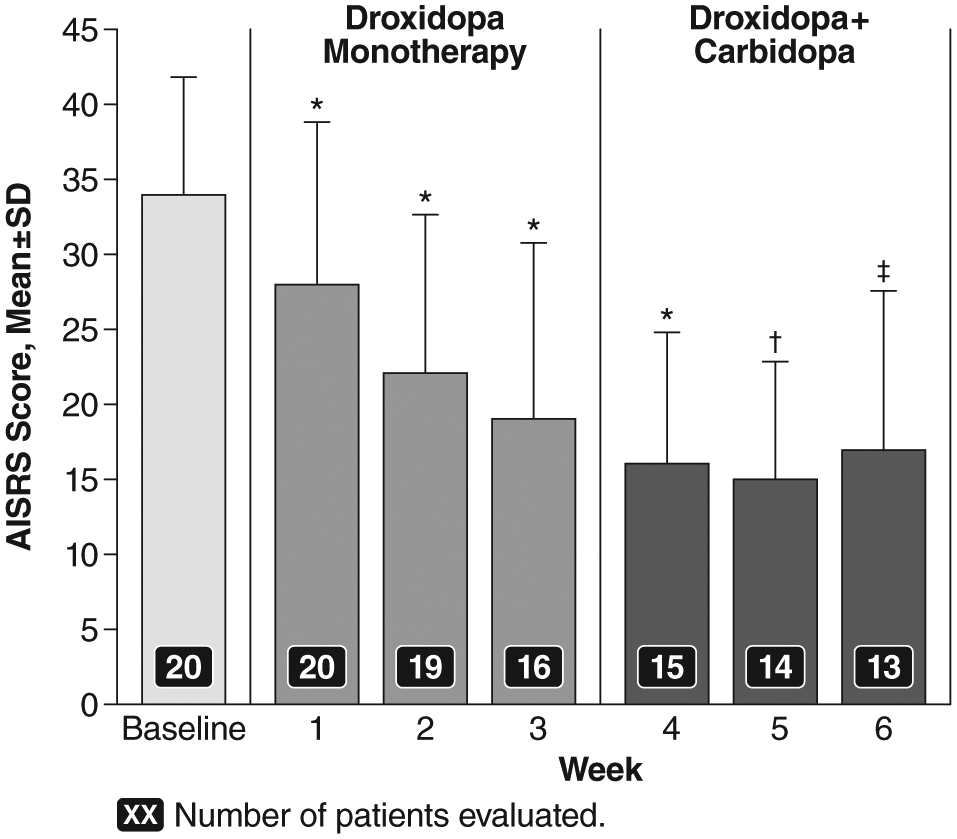
AISRS total scores (M ± SD) over time during the open-label, fixed-dose treatment period.
Total AISRS scores seen in the overall open-label period were maintained in both the droxidopa/carbidopa and placebo groups during the 2-week double-blind period. At Week 8, the mean ± SD total AISRS score was 19 ± 11.2 (mean change from baseline: −17 ± 10.4) in the droxidopa/carbidopa group and 15 ± 7.6 (mean change from baseline: −17 ± 4.1) in the placebo group. The difference between the two groups was not statistically significant (p = .99).
Secondary Endpoint: AISRS Inattentive and Hyperactive/Impulsive Subset Scores
As with the primary endpoint, improvements in both the AISRS Inattentive and the Hyperactive/Impulsive subset scores were seen during open-label droxidopa titration (Figure 4). At baseline, the mean ± SD score was 19 ± 4.1 for the Inattentive subset and 15 ± 3.9 for the Hyperactive/Impulsive subset. At Week 1, the mean Inattentive subset score was16 ± 6.4 (mean change from baseline: −3 ± 3.5; p = .0002) and the mean Hyperactive/Impulsive subset score was 12 ± 5.1 (mean change from baseline: −3 ± 3.1; p < .0001). At Week 3, the mean Inattentive subset score was 11 ± 7.4 (mean change from baseline: −8 ± 5.2; p = .0002) and the mean Hyperactive/Impulsive subset score was 8 ± 5.3 (mean change from baseline: −7 ± 3.7; p < .0001). The reduction in AISRS subset scores from baseline did not further improve with the addition of open-label carbidopa (25 or 50 mg), with a mean Inattentive subset score of 10 ± 6.2 (mean change from baseline: −9 ± 4.3; p = .0005) and a mean Hyperactive/Impulsive subset score of 7 ± 4.7 (mean change from baseline: −9 ± 3.1; p = .0002) at Week 6.
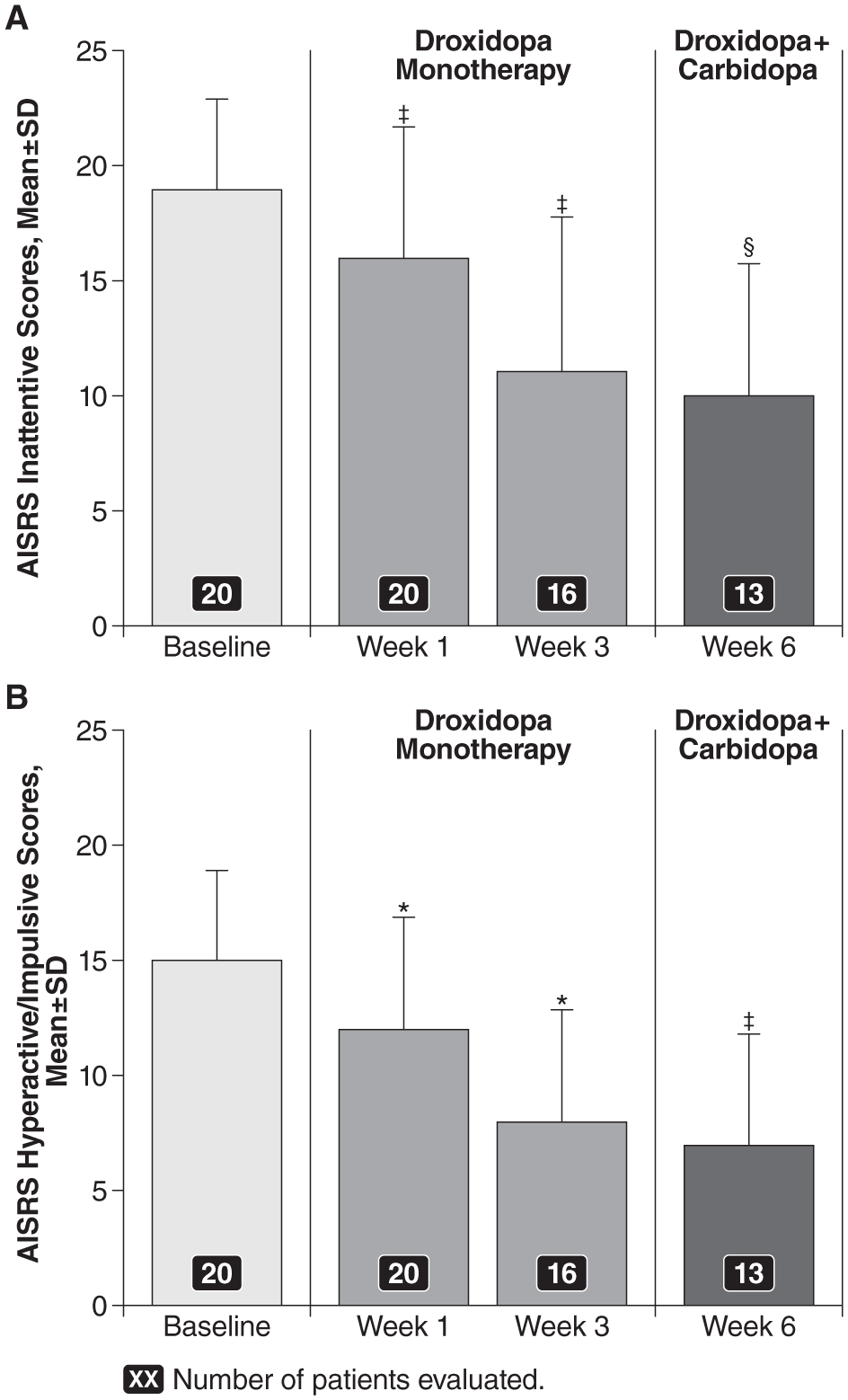
AISRS subset scores (M ± SD) for the (A) inattentive subset and (B) hyperactive/impulsive subset over time during the open-label, fixed-dose treatment period.
AISRS subset scores were maintained in both the droxidopa/carbidopa and the placebo groups during the 2-week double-blind period. At Week 8, the mean ± SD Inattentive subset score was 12 ± 6.1 (mean change from baseline: −8 ± 5.9) in the droxidopa/carbidopa group and 10 ± 5.3 (mean change from baseline: −8 ± 2.9) in the placebo group. The difference between the two groups was not statistically significant (p = .96). The mean Hyperactive/Impulsive subset score at Week 8 was 7 ± 5.3 (mean change from baseline: −9 ± 4.8) in the droxidopa/carbidopa group and 5 ± 3.7 (mean change from baseline: −9 ± 3.2) in the placebo group. The difference between the two groups was not statistically significant (p = .95).
Secondary Endpoint: ASRS Total and Subset Scores
Consistent with the improvements in AISRS scores, a significant reduction in mean ± SD ASRS total scores from a mean baseline score of 46 ± 9.4 was detected as early as 1 week after droxidopa treatment (42 ± 11.4; mean change from baseline: −4 ± 5.7; p = .0019). The mean ASRS total score decreased to 35 ± 10.9 (mean change from baseline: −11 ± 7.4; p < .0001) at Week 3. With the addition of open-label carbidopa (25 or 50 mg), the mean total score was 32 ± 11.2 at Week 6 (mean change from baseline: 15 ± 8.0; p = .0005). The improvements in total ASRS scores were maintained in both the droxidopa/carbidopa and the placebo groups during the 2-week double-blind treatment period, with a mean total score of 31 ± 14.9 (mean change from baseline: −15 ± 5.1) in the droxidopa/carbidopa group and 25 ± 8.0 (mean change from baseline: −19 ± 8.6) in the placebo group. The between-group differences were not significant (p = .39). Similar results were seen for the Inattentive and Hyperactive/Impulsive subset scores of the ASRS (data not shown).
Secondary Endpoint: CGI
Patients enrolled in the study reported marked functional impairment at baseline (mean ± SD CGI score of 5 ± 0.7). Within 1 week of initiating droxidopa monotherapy, the mean CGI score fell to 4 ± 1.0 (mean change from baseline: −1 ± 0.8; p = .0176), a level of moderate functional impairment. Improvement was maintained throughout the open-label treatment period, with a mean CGI score at Week 6 of 3 ± 0.9 (mean change from baseline: −1 ± 0.8; p = .0010), consistent with mild functional impairment. At Week 8, after the 2-week double-blind period, the mean CGI score was 4 ± 1.0 (mean change from baseline: −1 ± 1.2) in the droxidopa/carbidopa group and 3 ± 1.0 (mean change from baseline: −1 ± 0.9) in the placebo group. The difference between groups was not statistically significant.
Safety
The majority (90%) of participants reported at least one AE during treatment (Table 2). AEs were reported by 70% of patients during open-label droxidopa monotherapy and by 63% of patients during open-label droxidopa/carbidopa therapy. In the double-blind period, 67% of those in the droxidopa/carbidopa group and 40% of those in the placebo group reported AEs.
AEs in the Safety Population Present in ≥5% of Patients.
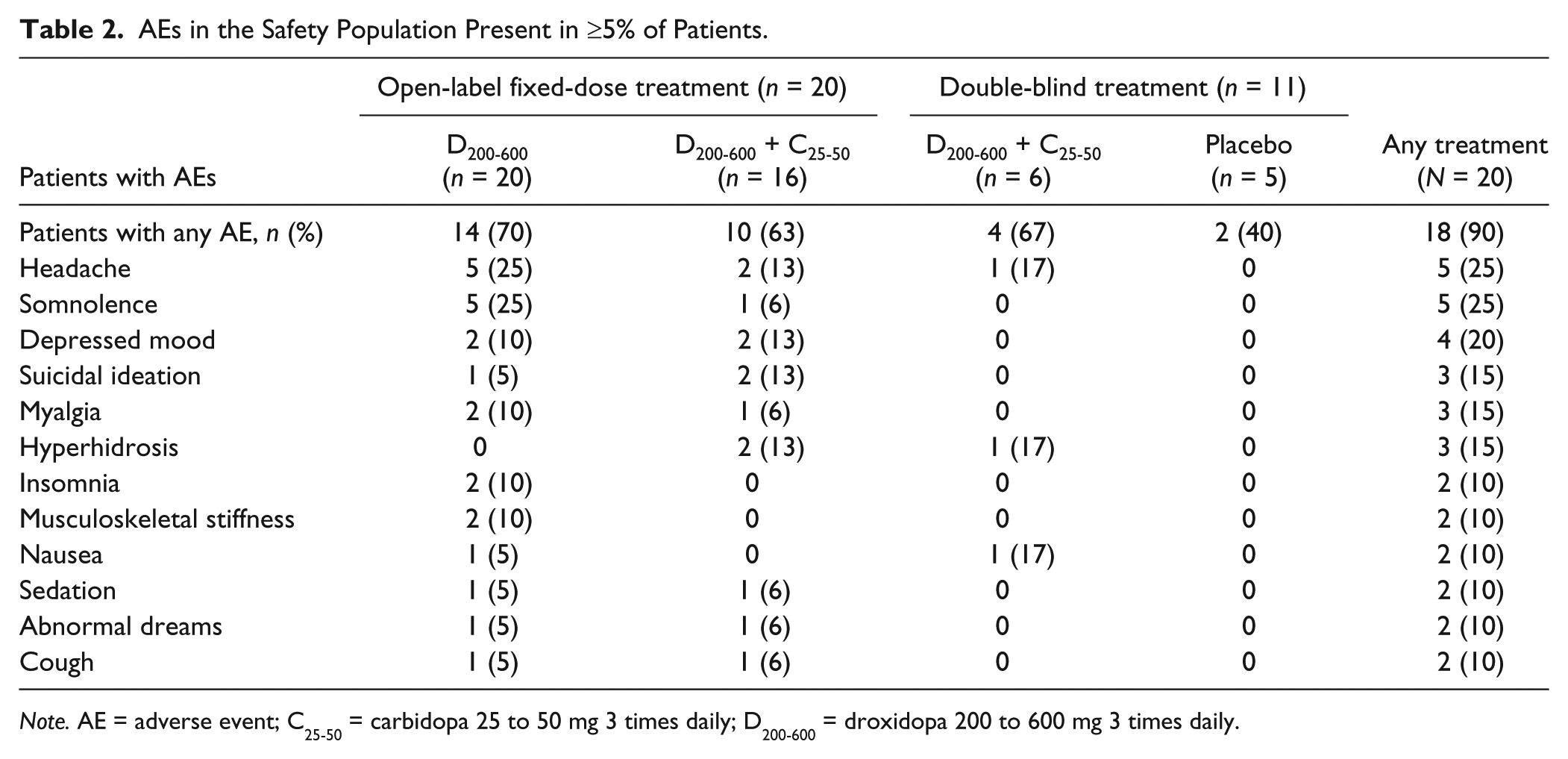
Note. AE = adverse event; C25-50 = carbidopa 25 to 50 mg 3 times daily; D200-600 = droxidopa 200 to 600 mg 3 times daily.
The intensity of AEs was predominantly mild in 55% of patients and moderate in 30% of patients, with one patient reporting two severe AEs (suicidal ideation and violence-related ideation) during open-label droxidopa/carbidopa therapy (Table 3). AEs were considered study-drug-related in the majority (80%) of cases. None of the AEs were SAEs. The majority (91%) of AEs were resolved by the end of the 30-day follow-up period.
Patients Reporting AEs in the Safety Population.
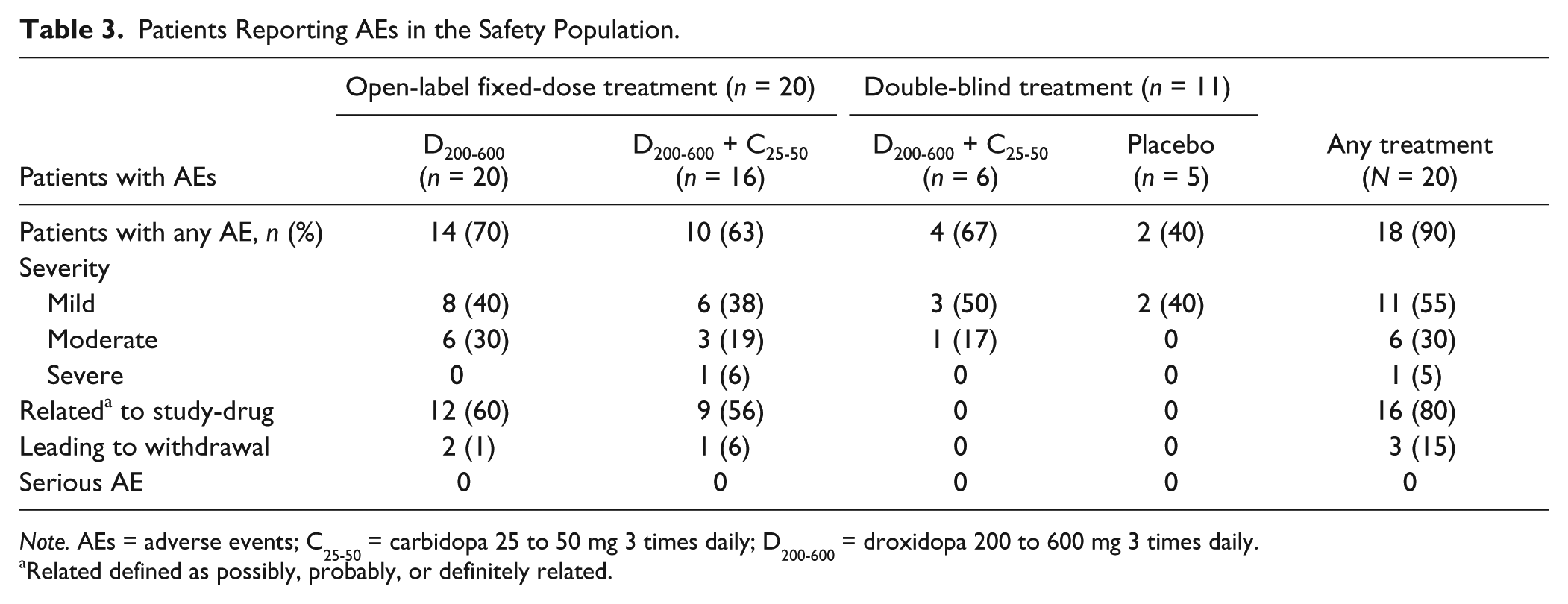
Note. AEs = adverse events; C25-50 = carbidopa 25 to 50 mg 3 times daily; D200-600 = droxidopa 200 to 600 mg 3 times daily.
Related defined as possibly, probably, or definitely related.
ECG abnormalities were reported for eight patients. In most cases, the abnormalities were limited to a single visit and the majority of the events were considered clinically non-significant. ECG abnormalities were reported as AEs and led to treatment and study discontinuation in two patients as described above.
There were no significant increases from baseline in systolic and diastolic blood pressure during the open-label period. The mean ± SD systolic blood pressure was 117 ± 10.4 at baseline and 115 ± 10.3 at Week 6. The mean diastolic blood pressure was 73 ± 9.2 at baseline and 74 ± 9.9 at Week 6. None of these differences were statistically significant. In the double-blind period, the mean systolic blood pressure was 114 ± 14.5 in the droxidopa/carbidopa group and 120 ± 10.7 in the placebo group at Week 7. The mean diastolic blood pressure at Week 7 was 69 ± 10.5 in the droxidopa/carbidopa group and 72 ± 5.5 in the placebo group. At Week 8, the mean systolic blood pressure was 122 ± 7.8 in the droxidopa/carbidopa group and 128 ± 11.1 in the placebo group. The mean diastolic blood pressure was 69 ± 8.2 in the droxidopa/carbidopa group and 76 ± 11.0 in the placebo group at Week 8. These differences were not statistically significant.
There were no significant differences in mean ± SD heart rate throughout the open-label period. Mean heart rate was 76 ± 12.8 at baseline and 76 ± 15.2 at Week 6. In the double-blind period, the mean heart rate was 81 ± 9.8 in the droxidopa/carbidopa group and 76 ± 19.0 in the placebo group at Week 7. At Week 8, the mean heart rate was 86 ± 8.4 in the droxidopa/carbidopa group and 68 ± 10.9 in the placebo group; none of the differences at Week 7 or 8 between the droxidopa/carbidopa and placebo groups were statistically significant.
There were no clinically meaningful changes in hematology, blood chemistry parameters, or mean body weight during the study.
Adherence
Study-drug adherence was monitored by regular capsule counts, and 80% adherence was pre-defined as acceptable for this study. The percentage of patients who met the acceptable adherence threshold declined over time, from 95% at Week 1 to ≤50% by the end of the open-label period. During the 2-week double-blind period, the percentage of patients who met the acceptable adherence threshold was 67% at Week 7 and 75% at Week 8 in the droxidopa/carbidopa group, and it was 60% at Week 7 and 25% at Week 8 in the placebo group.
Discussion
This proof-of-principle, pilot study demonstrated a statistically and clinically significant treatment effect of droxidopa administered as monotherapy and in combination with carbidopa in adult ADHD. Significant reductions from baseline in the primary endpoint of mean total AISRS score were seen at Week 1. Further significant improvements were documented at the end of the 3-week, open-label droxidopa titration period. The mean scores stabilized during 3 weeks of fixed-dose droxidopa plus carbidopa therapy; the addition of carbidopa did not improve mean scores above that seen at the end of dose titration with droxidopa monotherapy. Improvements from the open-label period were maintained in the double-blind period, and no significant between-group differences were seen between the droxidopa/carbidopa treatment group and the placebo group at Week 8.
As with the primary endpoint, there were statistically significant improvements in secondary endpoint mean scores after 1 week of droxidopa monotherapy, including mean AISRS Inattentive and Hyperactive/Impulsive subset scores, mean total ASRS score, and mean ASRS Inattentive and Hyperactive/Impulsive subset scores. These initial improvements were sustained throughout the remainder of the 3-week, open-label droxidopa titration period and after 3 weeks of fixed-dose droxidopa plus carbidopa therapy, although again, further gains were not made with the addition of carbidopa. There were no statistically significant between-group differences at the end of the double-blind treatment period for any of the secondary endpoints.
Functional impairment was improved by treatment with droxidopa or droxidopa/carbidopa. At baseline, the mean CGI score of 5 indicated marked functional impairment. Three weeks of droxidopa monotherapy resulted in a significant improvement to a mean score of 3 (mild functional impairment). Again, no further improvements in CGI score were seen with the addition of carbidopa, but scores were maintained throughout open-label combination treatment and during the double-blind treatment period.
This study was designed in such a way as to detect an additive benefit of carbidopa on the efficacy endpoints when initiated at Week 3 along with droxidopa. We hypothesized that inhibition of peripheral DDC through carbidopa co-treatment would increase CNS levels of NE. Indeed, in patients with Parkinson’s disease, levodopa is commonly given with carbidopa to reduce peripheral conversion, minimizing side effects and maximizing the amount of levodopa crossing the BBB (Marsden, Parkes, & Rees, 1973). The lack of significant benefit with regard to efficacy was unexpected. Carbidopa may still have contributed to safety by reducing the potential for peripherally mediated increases in blood pressure. No statistically significant changes in blood pressure or heart rate were seen at any point during study. Further studies are warranted to investigate the potential for combination therapy to improve the safety profile of droxidopa treatment for adult ADHD.
It was also unexpected that no significant differences in efficacy measurements were seen between the droxidopa/carbidopa group and the placebo group after the 2-week double-blind period of the trial. One likely explanation is that patients in the placebo group continued to experience the therapeutic benefits of prolonged droxidopa treatment, even after its discontinuation, for the short, 2-week washout period. A recent crossover study of lisdexamfetamine versus mixed amphetamine salts found that even for psychostimulants, short 1- to 2-week placebo washouts may not be sufficient for ADHD scores to return to baseline from the initial placebo treatment (Adler, Alperin, Leon, & Faraone, 2014). Further confounding factors may be the small number of patients randomized to double-blind treatment, with six patients receiving droxidopa/carbidopa and five patients receiving placebo, as well as low adherence with the treatment regimen. Only three patients in the combination treatment group and one patient in the placebo group were adherent during the second week of double-blind study treatment, a weakness in the present study. The relatively low adherence rate may have resulted from the extended nature of the open-label period (8 weeks) or the patient selection for individuals willing to enter a pilot study with a relatively complicated design. AEs, discussed below, may also have affected adherence rates.
A high percentage of patients experienced AEs in this study, although the intensity of the AEs was mainly mild-to-moderate. The most frequently reported AEs, such as headache (25%) and somnolence (25%), are consistent with the known safety profile of droxidopa (NORTHERA, 2014). Depressed mood was reported by four patients, and suicidal ideation was reported by three patients. Only one of these patients discontinued the trial; the other patients’ symptoms were short lived and resolved spontaneously. It is difficult to fully assess whether these events were related to the study-drug, as there was no placebo control comparison arm, and because there is some evidence that adult ADHD patients are at increased risk of experiencing suicidal ideation (Furczyk & Thome, 2014). Of note, droxidopa monotherapy for neurogenic orthostatic hypotension has not been associated with depression or suicidal ideation (NORTHERA, 2014). For Parkinson’s disease, the combination of levodopa plus carbidopa has been associated with a risk of depression, suicidal ideation, abnormal thinking and behavior, and aggression (SINEMET, 2014). Further studies of the combination of droxidopa plus carbidopa in adult patients with ADHD should be conducted with careful attention to patient selection and with increased use of formal assessments of suicidality.
In summary, findings from this proof-of-concept, pilot study suggest that droxidopa can improve the symptoms of adult ADHD. There may have been some participant selection bias due to the fact that this was a heavily monitored pilot study. Nevertheless, the safety profile was consistent with those reported previously, although the incidence of depressed mood in four patients and suicidal ideation in one patient was concerning. Future studies will be needed to determine whether the suicidality was primarily due to ADHD or whether it was related to the study medications. Future studies should also prospectively follow for ECG changes seen in several of the patients, which spontaneously resolved after medication discontinuation and have not been consistently reported in prior trials of droxidopa. Although this was a pilot study, the high drop-out rate also needs to be evaluated in future studies in terms of its influence on study results and the need to perform a modified-ITT analysis to account for these dropouts. These preliminary findings should be viewed with some caution because of the small sample size and open-label nature of treatment; however, they provide a plausible scientific rationale for additional studies of droxidopa in adult ADHD with larger samples and under more controlled conditions.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Lenard A. Adler, MD, has been a consultant and on the advisory board to Alcobra Ltd., Shire Pharmaceuticals, Theravance, the National Football League, and Major League Baseball. Dr. Adler has also received royalty payments (as inventor) from New York University for license of adult ADHD scales and training materials since 2004. Stephen W. Gorny, MS, is an employee of Lundbeck NA Ltd., Charlotte, NC.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this study was provided by Chelsea Therapeutics, Inc. (NCT#00983814), which was acquired in 2014 by Lundbeck LLC. Editorial assistance, which was financially supported by Lundbeck NA LLC, was provided by Jillian Lokere, MS, of The Curry Rockefeller Group, LLC, Tarrytown, NY. Lenard A. Adler has received research and grant support from Shire Pharmaceuticals, Chelsea Therapeutics, Inc. (now known as Lundbeck NA Ltd), Department of Veterans Affairs Cooperative Studies, Theravance, and APSARD/Pont Foundation. Investigator initiated grant from Chelsea Therapeutics to NYU School of Medicine and not directly to Lenard A. Adler, MD.