
Abstract
Proteases are industrially important enzymes but often have to be improved for their catalytic efficiency and stabilities to suit applications. Flow cytometry screening technology based on in vitro compartmentalization in double emulsion had been developed and applied on directed evolution of paraoxonase and β-galactosidase. Further advancements of flow cytometry–based screening technologies will enable an ultra-high throughput of variants offering novel opportunities in directed enzyme evolution under high mutational loads. For the industrially important enzyme class of proteases, a first flow cytometry–based screening system for directed protease evolution has been developed based on an extracellular protease-deficient Bacillus subtilis strain (WB800N), a model protease (subtilisin Carlsberg), and a water-in-oil-in-water double-emulsion technology. B. subtilis WB800N cells are encapsulated in double emulsion with a fluorogenic substrate (rhodamine 110–containing peptide), allowing the screening of protease variants in femtoliter compartments at high throughput. The protease screening technology was validated by employing an epPCR mutant library with a high mutational load and screened for increased resistance toward the inhibitor antipain dihydrochloride. A variant (K127R, T237P, M239I, I269V, Y310F, I372V) with an improved relative resistance was isolated from a small population of active variants, validating the reported protease flow cytometry screening technology for increased inhibitor resistance.
Introduction
P
Directed protease evolution campaigns mainly use traditional screening formats such as agar plate (halo formation) or microplate-based detection systems. 12 The agar plate assays are usually applied for a prescreening to identify the proteolytic activities at simple reaction conditions. The microplate-based screening methods are commonly used in directed evolution with capacities to be applied for more complex reaction cases. The two systems are usually associated with low mutational loads (one to three amino acid changes per mutated gene) so that a medium throughput is sufficient to find improved variants. 13 A notable example for a high-throughput screening (HTS) of protease variants is the directed evolution of the surface membrane protease OmpT to alter its substrate specificities by employing an Escherichia coli OmpT-deficient strain (UT5600). 14 Improved variants were identified using flow cytometry in aqueous solution without employing any compartmentalization technology.
Recently, a (water-in-oil-in-water [w/o/w]) double-emulsion-based compartmentalized flow cytometry screening technology (in vitro compartmentalization [IVC]) has emerged as a potentially powerful screening format for sampling mutant libraries with approximately up to 108 variants per day. 15 The IVC systems use a manmade cell compartmentalization (e.g., w/o/w double-emulsion droplets), in which enzymatic reactions can be performed individually in femtoliter volume scale, resulting in a significant decrease of consumable cost. 16 The principle of IVC-based flow cytometry screening systems employs fluorescence detection of a fluorescent product derived from a fluorogenic substrate. Therefore, the existence of a retainable fluorescence signal and its intensity are key parameters for the successful implementation of flow cytometry analysis employing IVC technologies. 17 In contrast to the surface adsorption method, the compartmentalized screening technology (IVC) is more generally applicable and could enable novel evolution strategies, for instance, by employing libraries with high mutational loads. Progress in IVC technology has been summarized well in several reviews, 18–20 and the following screening systems have been reported in directed evolution of paraoxonase 21 and β-galactosidase. 22 Methods based on selection systems with or without the application of IVC technologies are always extremely powerful as the survival of an organism is coupled to the desired protease function, reducing the necessary workload by far. Such selection systems have been developed for screening trypsin using minimal media supplemented with arginyl naphthylamide. 23 The mentioned selection systems are limited in their general applicability as suitable host strains have to be generated for growth selection.
Here we report a first flow cytometry screening system based on IVC in w/o/w double emulsion for the directed evolution of proteases (ProFC-IVC), with an overview on the flow cytometer–based protease screening technology, including the main steps and screening procedures. The ProFC-IVC screening system was validated for directed evolution of subtilisin Carlsberg (SC) toward increased inhibitor resistance (antipain dihydrochloride) employing an epPCR mutant library with a high mutational load.
Materials and Methods
Materials
All chemicals used were of analytical reagent grade or higher quality and purchased from Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany), Applichem (Darmstadt, Germany), or Carl Roth (Karlsruhe, Germany). All enzymes were purchased from New England Biolabs (Frankfurt, Germany), Fermentas GmbH (St. Leon-Rot, Germany), Sigma-Aldrich, or Codexis (Redwood City, CA). Fluorogenic substrate bis, Succinyl-L-Ala-L-Ala-L-Pro-L-Phe-rhodamine 110 (bis, Suc-AAPF-R110), was synthesized by AnaSpec (San Jose, CA), and protease reversible inhibitor antipain dihydrochloride (antipain-HCl) was purchased from Sigma-Aldrich. Plasmid isolation kit, PCR purification kit, and rolling circle amplification kit (REPLI-g) were from Qiagen (Hilden, Germany).
All PCRs were performed using a thermal cycler (Mastercycler gradient; Eppendorf AG, Hamburg, Germany). The amount of DNA in the experiments was quantified by using a NanoDrop photometer (ND-1000; NanoDrop Technologies, Wilmington, DE). Fluorescence was measured by using a fluorescence microplate reader (Tecan SAFIRE, Tecan Austria GmbH, Salzburg, Austria; excitation 488 nm, emission 520 nm) or a flow cytometer (CyFlow space; Partec GmbH, Muenster, Germany).
Bacteria, plasmids, and cultivation conditions
E. coli DH5α (Stratagene, La Jolla, CA) and E. coli DH10B (E-Shot DH10B T1R Electrocomp Cells; Invitrogen GmbH, Darmstadt, Germany) were used as cloning hosts. Bacillus strains were used for the expression and generation of protease libraries. B. subtilis WB600 was provided by the collaborator B.R.A.I.N. AG (Zwingenberg, Germany), and B. subtilis WB800N 24 carrying neomycin resistance was kindly provided by Prof. Wolfgang Schuman. Plasmids pBCS (shuttle vector) and pBCSSC (pBCS carrying the subtilisin Carlsberg gene) were provided by B.R.A.I.N. AG.
Cells were routinely cultivated in Luria broth (LB) supplemented with appropriate antibiotics using a shaking incubator (Multitron II; Infors GmbH, Einsbach, Germany) at 37°C and 250 rpm for 20 h. Antibiotics used in the experiment were chloramphenicol (Cm, 12.5 µg/mL) and neomycin (Ne, 10 µg/mL).
Assays for the detection of protease activity
Agar plate assay
The protease-secreting Bacillus strains were cultivated on LB agar plates supplemented with 1% (w/v) skim milk, Cm 12.5 µg/mL, and Ne 10 µg/mL, and protease activity was detected by halo formation.
Fluorimetric microplate assay
The assay was carried out in 384-well microplates (polystyrene black flat 384-well no. 3710; Corning, Inc., Corning, NY). Reactions were performed in Tris-HCl buffer (0.1 M, pH 8.6) containing the fluorogenic substrate bis, Suc-AAPF-R110 (5 µM), and cell supernatant (1 µL) in the presence or absence of the reversible inhibitor antipain-HCl (1 µM) in a final volume of 50 µL. The enzyme activity was measured at 37°C by monitoring the increase of fluorescence intensity using the Tecan SAFIRE.
Normalization of protein concentrations for wild-type SC and variants. Protein concentrations in fluorescence assays were normalized by using the Agilent 2100 Bioanalyzer (Agilent Protein 230 Kit; Agilent Technologies, Böblingen, Germany) and the BCA protein assay kit (no. 23225; Pierce Biotechnology, Rockford, IL) according to the manual’s instructions.
Generation of the epPCR mutagenesis library of subtilisin Carlsberg
The random mutagenesis library was constructed by standard epPCR 25 followed by megaprimer PCR of whole plasmid (MEGAWHOP) 26 as described. The epPCR library was generated by PCR (95°C for 5 min, 1 cycle; 94°C for 30 s/55°C for 30 s/72°C for 90 s, 30 cycles; 72°C for 7 min, 1 cycle) using dNTP mix (0.2 mM), primers (BLSCf1: 5′-ATGATGAGGAAAAAGAGTTTT-3′; BLSCr1: 5′-AGCGGCAGCTTCGACATTG-3′; 0.4 µM each), plasmid DNA template (pBCSSC, 10 ng), Taq polymerase (2.5 U), and MnCl2 (0.4 mM) in a final volume of 50 µL. The MEGAWHOP PCR (68°C for 5 min, 1 cycle; 95°C for 2 min, 1 cycle; 95°C for 30 s/55°C for 30 s/68°C for 12 min, 18 cycles; 68°C for 30 min, 1 cycle) was performed in a final volume of 50 µL using dNTP mix (0.2 mM), megaprimer (purified epPCR amplification product, 400 ng), plasmid DNA template (pBCSSC, 50 ng) and Pfu polymerase (2.5 U). The amplified MEGAWHOP product was digested by DpnI (20 U) at 37°C for 2 h, purified using the PCR purification kit (Qiagen), and transformed into E. coli strains. Plasmids were then pooled, isolated, and, after rolling circle amplification (REPLI-g, Qiagen), transformed into B. subtilis strains for expression and screening.
Emulsification of Bacillus cells
The w/o/w double emulsions were prepared as described previously. 15 Bacillus cultures were harvested using a bench centrifuge (Eppendorf centrifuge 5414D; Eppendorf AG, Hamburg, Germany) at 8000 g for 3 min and rinsed three times using ice-cold phosphate-buffered saline (PBS). Approximately 5 × 109 cells were suspended in 1 mL of Tris-HCl buffer (0.1 M, pH 8.6) and passed through a 8-µm syringe filter membrane (PC MB 25 mm 8.0 µm; Whatman GmbH, Dassel, Germany) prior to emulsification. For B. subtilis host cell selection, the cell suspension (~2 × 108 cells) was emulsified into w/o/w double emulsions together with the fluorogenic substrate bis, Suc-AAPF-R110 (50 µM), and the internal dye 7-hydroxycoumarin-3-carboxylic acid (no. 55155, Sigma-Alrich; 50 µM) and incubated at 37°C for 2–3 h (BD 115; BINDER, Tuttlingen, Germany). For epPCR mutant library screening, the emulsification condition was used as above in the presence of protease inhibitor (antipain-HCl, 1 µM).
Flow cytometry–based screening and sorting
Double emulsions were diluted 100-fold using PBS buffer and analyzed by using the CyFlow space (Partec GmbH; sheath fluid was composed of 0.9% [w/w] of NaCl and 0.01% [v/w] of Triton X-100). Fluorescence intensities of the rhodamine 110 (R110) products (green fluorescence) and the internal dye (blue fluorescence) were recorded, and the sort gates were set to collect events with control blue fluorescence (internal dye) and increased green fluorescence (R110 products). The sorting rate was controlled to 5–10 events/s using 8000 events/s analysis speed, and around 104 events were sorted into a 50-mL solution. The sorted samples were then recovered by filtration (no. 7182-009; Whatman GmbH, Dassel, Germany) followed by incubation on LB plates with the filter membrane upwards placed. Grown cells were further scraped from the membrane and subjected to the next round of screening.
Single amino acid substitution and computational analysis of mutant variants
Single amino acid site-directed mutagenesis (SDM) of the SCm2 variant was performed according to the published method. 27 The sequences of the oligonucleotides used to create the six individual mutant variants SCm2 are as follows, where triplets at the substituted site are underlined: K127R (K127Rf 5′-CAGGCTCAAGGCTTTAGGGGAGCGAATGTAAAA-3′; K127Rr 5′-TTTTACATTCGCTCCCCTAAAGCCTTGAGCCTG-3′), T237P (T237Pf 5′-GGAGCATCAGGCTCGCCAGCGATGAAACAGGCA-3′; T237Pr 5′-TGCCTGTTTCATCGCTGGCGAGCCTGATGCTCC-3′), M239I (M239If 5′-TCAGGCTCGACAGCGATAAAACAGGCAGTCGAC-3′; M239Ir 5′-GTCGACTGCCTGTTTTATCGCTGTCGAGCCTGA-3′), I269V (I269Vf 5′-GGAAACACGAATACAGTTGGCTATCCTGCGAAA-3′; I269Vr 5′-TTTCGCAGGATAGCCAACTGTATTCGTGTTTCC-3′), Y310F (Y310Ff 5′-CCTGGCGCAGGCGTATTCAGCACTTACCCAACG-3′; Y310Fr 5′-CGTTGGGTAAGTGCTGAATACGCCTGCGCCAGG-3′), and I372V (I372Vf 5′-TATGGGAAAGGTCTGGTCAATGTCGAAGCTGCC-3′; I372Vr 5′-GGCAGCTTCGACATTGACCAGACCTTTCCCATA-3′). Molecular modeling structures were generated by SWISS-MODEL SIB Service (http://swissmodel.expasy.org/) and analyzed by using the Visual Molecular Dynamics software (http://www.ks.uiuc.edu/Research/vmd/).
Results
The protease flow cytometry screening system (ProFC-IVC) was developed by employing subtilisin Carlsberg as a model protease. Optimized parameters comprised (a) substrate selection to avoid partitioning of substrate and product into the oil phase, (b) host strain selection to minimize protease background reactions, and (c) host strain recovery after sorting. The developed protocol was subsequently validated by screening an epPCR library in which the mutational load was adjusted to 2%–3% of active clones.
Substrate selection to avoid partitioning of substrate and product into the oil phase
A key parameter for a successful IVC screening protocol is to avoid cross-talk between individual compartments or a reduction of the fluorescence signal by diffusion of substrates and/or products into the surrounding medium. Figure 1 shows w/o/w double emulsion after 4 h of incubation where subtilisin Carlsberg from culture supernatants had been entrapped together with its fluorogenic substrate bis, Suc-AAPF-R110. The fluorescent reaction product (rhodamine 110) is solely located within the inner aqueous phase of the emulsion (labeled as w1), illustrating that the reaction only takes place inside the generated compartments. No considerable diffusion into the surrounding phase could be observed. The fluorescence signal was very stable, and its intensity did not show any influence by pH values between 7.0 and 9.0 and temperatures of 25°C and 37°C, respectively. Figure 2 shows the correlation between R110 product concentrations and the intensity of the detected fluorescence signal using the microplate assay ( Fig. 2a , for enzyme characterization in microplate) and flow cytometry analysis ( Fig. 2b , to define a sorting region for the library screening). The readout value of R110 fluorescence (as shown in Fig. 2a ) could be adjusted from 1–50 000 with different modulated amplifier stage gains (60–100).
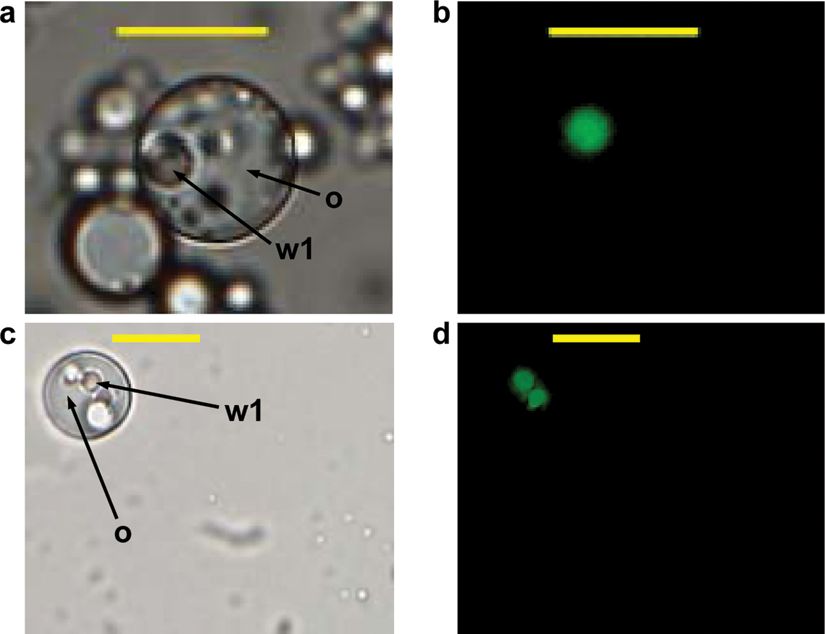
Fluorescence microscopy of double emulsions at light field (
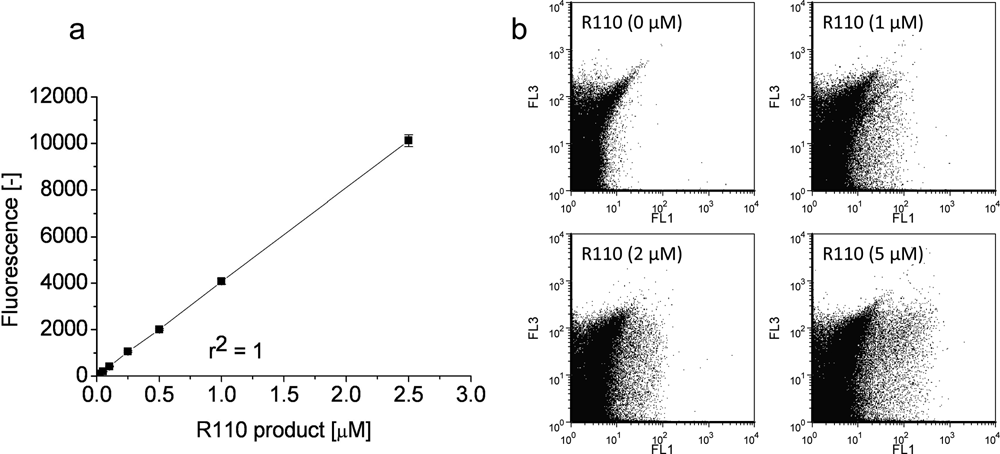
Correlation of fluorescence signal and rhodamine 110 (R110) concentration. The fluorescence intensity was determined using the Tecan SAFIRE microplate reader (excitation: 488 nm, emission: 520 nm, gain 70) in Tris-HCl (0.1 M, pH 8.6) at 37°C (
Host strain selection to minimize protease background reactions
During the procedure of host strain selection, first experiments with the selected substrate (bis, Suc-AAPF-R110) indicated that a lack of residual protease activity is an important prerequisite for an IVC screening technology for proteases. Therefore, background activities of two protease-deficient B. subtilis strains (WB600 and WB800N) were investigated in w/o/w double emulsions by flow cytometry. B. subtilis WB600 has six knockouts of extracellular proteases (nprE nprB aprE epr mpr bpr), and in B. subtilis WB800N, two additional proteases (vpr wprA) had been knocked out. Figure 3 shows the corresponding fluorescence signals of the released R110 product. Despite the knockouts, fluorescence signals could be clearly detected, proving the superb sensitivity of the ProFC-IVC detection system. In comparison, B. subtilis WB800N revealed a 6.7-fold lower background compared to B. subtilis WB600. The difference of signal intensities between protease-expressing cells and the empty vector control also proved to be by far more distinct for B. subtilis WB800N (26-fold) than for B. subtilis WB600 (3-fold). Figure 3c displays a visual illustration of the difference by overlaying samples in one FL1 histogram where the M1 marker represents the district for positive events. Because of the observed signal properties, B. subtilis WB800N proved to be a potentially suitable host for directed protease evolution, although exponential growth of this strain is ~2-fold slower than that of B. subtilis WB600.
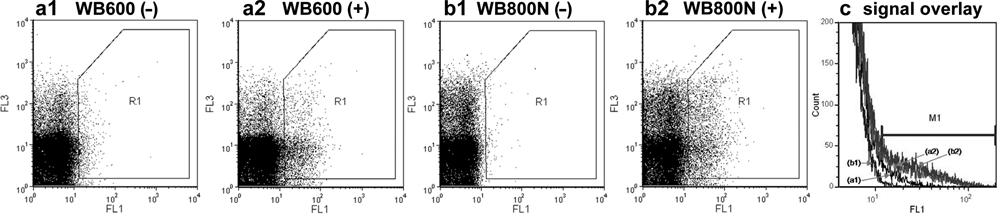
Flow cytometer recordings of fluorescence signals of protease-deficient Bacillus subtilis strains entrapped in water-in-oil-in-water (w/o/w) double emulsions. Strains harboring pBCSSC expressed subtilisin Carlsberg (+), whereas strains harboring pBCS served as negative controls (–). (
After host strain selection, the enzymatic reaction inside w/o/w double emulsions was further optimized concerning reaction buffer, pH, temperature, and reaction time. The results showed that alkaline pH and increased temperatures caused elevated enzyme activity, whereas cell viability was reduced significantly. Optimal reaction conditions for the aspired ProFC-IVC proved to be a slight alkaline pH of 8.6 (Tris-HCl buffer) at 37°C as they only led to marginal effects on cell viability, whereas a strong fluorescence signal could be obtained.
Host strain recovery after sorting
A major challenge using the ProFC-IVC system is the cultivation of cells after sorting, as well as keeping the enrichment in the population through iterative rounds of sorting. A loss of enrichments in sorted populations was observed if cells were regrown directly in liquid culture (3 mL LB in 15 mL of glass tubes). A rich inoculum (>105 cells) proved to be a prerequisite for preserving enrichment factors achieved in previous rounds of sorting. Because of this limitation, sorted variants (usually ~104 variants) were recovered first by vacuum filtration with a sterile membrane and subsequently grown on LB agar plates prior to cultivating in liquid media for the next round of sorting.
Mutant library generation and validation of ProFC-IVC
Three mutant libraries of subtilisin Carlsberg protease were generated by varying MnCl2 concentrations (0.1 mM, 0.2 mM, and 0.4 mM) with active ratios around 85%, 50%, and 2.3%, respectively. The high mutational load library (0.4 mM MnCl2 generating ~2.3% of active clones) was finally selected to generate the mutant library for screening. Bacillus strains usually display transformation rates <105 per µg DNA. To provide a library with an adequate size, a two-step transformation procedure as described in Materials and Methods was performed using E. coli for efficient subcloning. Individual colonies harboring protease mutants were pooled, and the corresponding plasmids were isolated and, after rolling circle amplification, transformed into B. subtilis WB800N. The diversity of the generated mutant library was approximately 105–106. The generated subtilisin Carlsberg variants were entrapped in a w/o/w double emulsion as described before. The number of emulsified aqueous droplets exceeded the number of entrapped cells by one to two orders of magnitude, ensuring entrapment of not more than one variant per double-emulsion droplet. The library was finally screened toward increased inhibitor resistance (antipain-HCl) to validate the developed ProFC-IVC screening system.
Our primary sorting experiments revealed “escapee” negative events present in the sorting channel, which can be attributed to the piezo activator sorter module of the Partec CyFlow space flow cytometer. A minimal sorting rate of 5–10 events/s at an analysis rate of ~8000 events/s using 100-fold diluted w/o/w double emulsions in PBS buffer was required to control this “leakage” in a low range. For each round of sorting, in total ~107 cells were analyzed using an input speed of 1–2 µL/s, and around 10 000 events with highest fluorescence signal were gated for sorting. After three iterative rounds of screening and sorting, the library could be enriched approximately 12-fold (active ratio was increased from ~2.5%–30%). Figure 4 shows the fluorescent signals of different sorting stages illustrating the achieved enrichment.
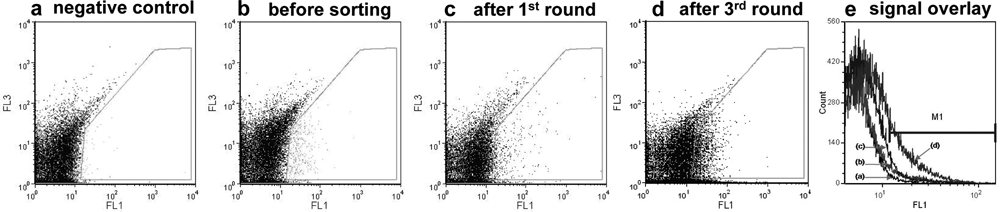
Flow cytometer recording of fluorescent signals of the compartmentalized subtilisin Carlsberg mutant library in Bacillus subtilis during different stages of sorting. (
Variants with increased inhibitor resistance and characterization
After the last round of sorting, a small fraction of the sorted cells (~1000 of ~10 000 collected events) was plated on LB skim milk agar plates, and from a total of 315 active colonies, 15 with the highest ratios of halo/colony size were selected for rescreening as a proof of concept of the protease flow cytometer screening system. From the rescreened variants, the clones SCm2 and SCm3 exhibited an increased residual activity in the presence of antipain-HCl (~2.1-fold and 1.2-fold, respectively) compared to the wild-type. Sequencing results revealed that variants SCm2 and SCm13 contained multiple mutations leading to amino acid substitutions ( Table 1 ). In addition, several silent mutations occurred on the DNA level of SCm2 (Y111, L136, P145, I279, A239) and SCm13 (S28, F69). Despite a reduced absolute activity of variants SCm2 and SCm13, improved inhibitor resistance for both variants was observed ( Table 1 ). The inhibitor improvement of SCm2 before purification was 213% (as shown in Table 1 ) and 160% after purification and normalization to the expression. Compared to a rather low standard deviation (3%–5%) of the microplate Suc-AAPF-R110 rescreening system, the identified variant SCm2 has a resistance that is outside of the standard deviation.
Figure 5 shows the positions of the obtained amino acid substitutions of variant SCm2. The corresponding six residues in the wild-type enzyme were further substituted individually by SDM to explore the contribution of each mutated position to the observed resistance property. In all assays, SC concentrations were normalized to avoid false results due to differential expression of the analyzed variants. Except for variant SCm2SDM3 (M239I), which exhibited a 1.2-fold increased inhibitor resistance compared to the wild-type enzyme, all mutants with single amino acid substitutions did not show any improvements so that no additive effects could be observed. After normalization, the SCm2 variant (K127R, T237P, M239I, I269V, Y310F, I372V) still proved to display a higher (1.6-fold compared to the wild-type) inhibitor resistance than the only improved single mutant.
Inhibitor Resistance and Amino Acid Changes of ProFC-IVC Sorted Variants of Subtilisin Carlsberg (SC) Compared to the Wild-Type Enzyme
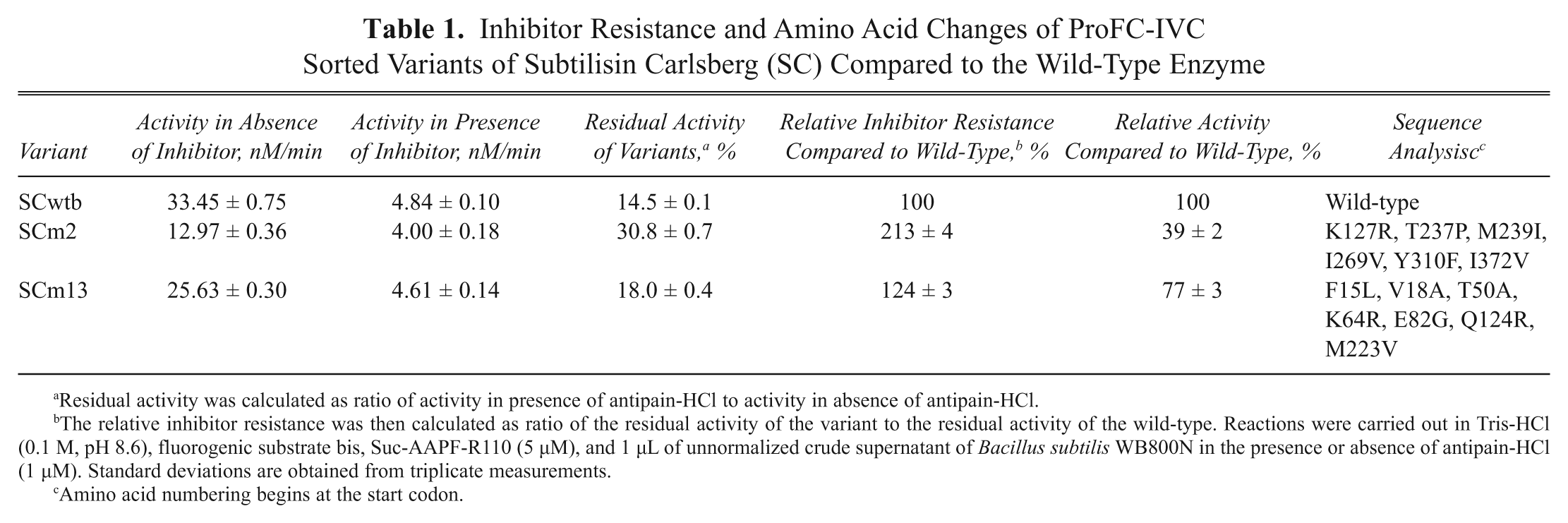
Residual activity was calculated as ratio of activity in presence of antipain-HCl to activity in absence of antipain-HCl.
The relative inhibitor resistance was then calculated as ratio of the residual activity of the variant to the residual activity of the wild-type. Reactions were carried out in Tris-HCl (0.1 M, pH 8.6), fluorogenic substrate bis, Suc-AAPF-R110 (5 µM), and 1 µL of unnormalized crude supernatant of Bacillus subtilis WB800N in the presence or absence of antipain-HCl (1 µM). Standard deviations are obtained from triplicate measurements.
Amino acid numbering begins at the start codon.
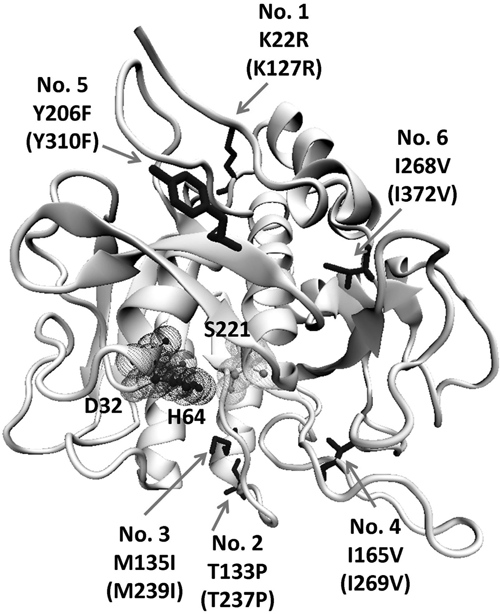
Positions of amino acid substitutions in mature subtilisin Carlsberg variant SCm2 displaying increased protease inhibitor resistance. Substitutions are indicated by arrows, and numbers in brackets specify the corresponding position counting from the start codon. D32, H64, and S221 form the catalytic triad in the active site of the protein. Discrepancies in numbering result from the numeration in the used model (1C3L) where all amino acids starting from position 56 are shifted by one position (missing position 56).
Discussion
Proteases are industrially important enzymes, and advances in screening technologies such as IVC offer novel opportunities to tailor protease properties by directed evolution. Main challenges in whole-cell IVC screening systems are substrate and host strain selection, cell recovery, and maintenance of achieved enrichments in sorted libraries. Figure 6 shows an overview of the developed ProFC-IVC screening technology summarizing the main screening procedure and solutions to the addressed challenges. The developed system employs a w/o/w IVC technology 17 with entrapment of protease-secreting B. subtilis cells in femtoliter reaction compartments. The w/o/w double-emulsion technology offers the opportunity to directly sort individual compartments using flow cytometry. 15
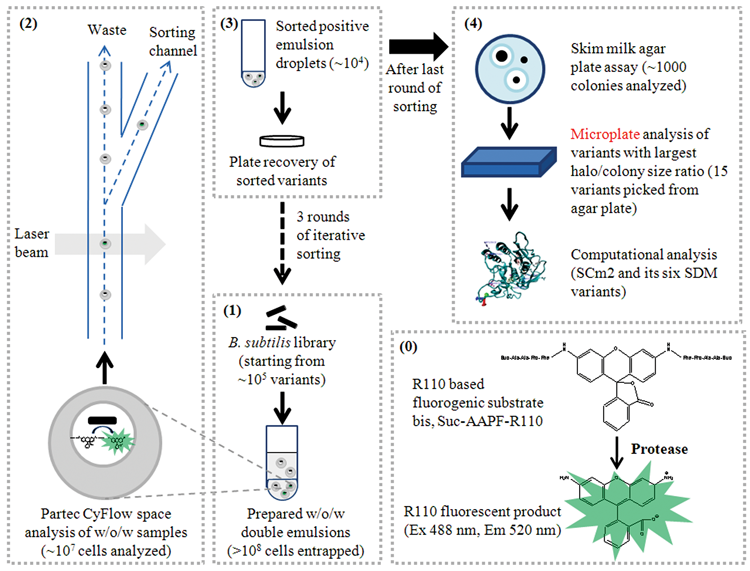
Scheme of the developed flow cytometry–based whole-cell screening system for proteases (ProFC-IVC). (
Rhodamine 110 is used in other reports for detecting protease activity, and for Suc-AAPF-R110, there is one report for determining kinetic constants in single water-in-oil emulsions and measuring hundreds of individual molecules simultaneously using a microscope and charge-coupled device camera. 28 For ProFC-IVC bis, Suc-AAPF-R110 ( Fig. 6(0) ) was selected as substrate for the model protease SC as the charges of its succinyl-group and its fluorescent hydrolysis product rhodamine 110 (R110) allow retention of substrate and product inside the compartments. The retention is a key factor for the intended screening because leakage of substrate and/or product would lead to a loss in signal intensity and to cross-talk between compartments, preventing the efficient sorting of individual events. Furthermore, the chosen substrate is not membrane permeable, avoiding additional background activities from intracellular proteases.
A second challenge to be addressed is the selection of a suitable host strain. Besides efficient heterologous protease expression, background activities must be minimized to avoid the selection of negative events during the sorting process. Because proteases occur naturally in all organisms, a system using secreted bacterial proteases was selected. B. subtilis was employed as expression host because of its naturally high secretion capacity as well as its safe and practical handling properties. 29 Our investigation of two extracellular protease-deficient B. subtilis strains showed that a strain where eight genes coding for extracellular proteases had been knocked out (B. subtilis WB800N) 24 was optimal for the intended application as it displayed the lowest background activities combined with high product fluorescence signals. In contrast, because of lower signal intensities and higher background, B. subtilis WB600 deficient for only six extracellular proteases proved to be less suitable.
Finally, growth competition was observed when cells were directly subjected to liquid cultivation after the sorting process. The biased growth led to a loss of enrichments, particularly after the first round of sorting, when the number of active cells in the inoculum was still low. Cell recovery by agar plate propagation prior to liquid cultivation could limit the effect of growth competition, thereby preserving the accomplished enrichments. Another approach to overcome the observed biased growth rates would involve the utilization of inducible expression systems. 30 However, the expression levels of SC using isopropyl-β-D-thiogalactopyranoside (IPTG) or xylose-inducible systems in B. subtilis 31 were reduced more than four-fold compared to the constitutive expression system. Consequently, an inducible expression was not considered for ProFC-IVC, and the additional plate cultivation step proved to be optimal to preserve enrichments in sorted libraries.
In our experiments, we used a flow cytometer with a piezoelectric activation sorter module because of the higher cell viability during the sorting process and its high signal resolution. Nevertheless, an unavoidable continuous leakage of fluid from the sorting channel ( Fig. 6 ) leads to the additional sorting of negative events, thereby decreasing achievable enrichment factors in each individual round. However, a high overall enrichment can be obtained by performing several iterative rounds of sorting.
The developed ProFC-IVC screening system was validated by screening an epPCR mutant library of SC with high mutational load (diversity of ~105; ~5 mutations per 1 kb) for variants with improved resistance toward the protease inhibitor antipain-HCl. The property inhibitor resistance was selected because of potential applications interest for liquid detergents and simplicity in handling. To avoid improving the activity for the specific substrate Suc-AAPF-R110 and reducing the activity toward complex protein stains, other properties (e.g., pH, temperature, peroxide) that are not related to degrade complex protein stains can also be applied for screening. A variant with six amino acid substitutions could be identified exhibiting a 160% increased resistance to the inhibitor compared to the wild-type. The obtained improvement after normalization is rather low as only a small fraction of the collected cells was rescreened for a conceptional proof of the flow cytometer screening system. Rescreening of a larger fraction of active clones or more stringent sorting conditions might likely lead to the isolation of protease variants with further improved properties. The mutated gene contained several silent mutations, possibly contributing to increased expression levels of this variant, which shows the necessity of normalizing protein concentrations for the characterization of the identified variants. The generated single-mutant experiments did not show any additive effects of the single mutations. Hence, double and triple mutations would have to be generated to investigate the likely cooperative effects of the identified amino acid changes to explain the improved inhibitor resistance.
In general, the high-throughput capacities of flow cytometry–based screening systems (>108 events per day) offer opportunities for alternative directed evolution strategies. In contrast to conventional microplate or agar plate assays where small libraries with low mutational loads can be screened with modest improvements and repeated rounds of mutation and screening, the high throughput of flow cytometry–based methods allows screening of large libraries with high mutational loads. 18 In our case, transformation efficiencies of B. subtilis restricted the size of the screened epPCR mutant library. However, by employing advanced transformation technologies to overcome the observed limitation, the developed ProFC-IVC screening system can be used to analyze also large libraries in a high throughput. This would allow the investigation of cooperative effects between multiple mutations, especially in terms of structure function illumination. The described whole-cell-based ProFC-IVC screening platform presents a powerful screening tool for engineering proteases by directed evolution. Nevertheless, its successful application demands the consideration of several limiting factors such as substrate selection, background activities of the employed host cells, transformation efficiencies, and cell recovery. 15 Most of the above-mentioned limitations might be overcome by utilization of efficient cell-free in vitro expression, which would allow harnessing fully the throughput capabilities of the developed ProFC-IVC screening technology.
In summary, the first use of flow cytometry to sort the cells in w/o/w double emulsions for directed protease evolution has been developed and analyzed by screening an epPCR library for inhibitor resistance of subtilisin Carlsberg. The reported screening system in Bacillus WB800N, due to its sensitivity, can likely be modified for other proteases and applied for additional properties such as high stability/activity at low and high temperature, altered pH profile, or ionic strength resistance.
Footnotes
Acknowledgements
We thank Milan Blanusa and Raluca Ostafe for valuable discussion, BMBF (program: BioChancePLUS-3 FKZ 0313137) for financial support, B.R.A.I.N. company (especially Dr. Jürgen Eck, Dr. Frank Niehaus, and Dr. Patrick Lorenz), and Partec Company (especially Dr. Danny Köhler) for technical support.