
Abstract
Nucleic acid drug delivery with lipid nanoparticle (LNP) formulations has enabled the development of novel therapeutics and vaccines. LNP formulations are composed of both naturally occurring and synthetic lipid excipients. This perspective shares current practices in the nonclinical safety assessment of novel lipid excipients contained in LNP formulations and identifies gaps in current regulatory guidance on this topic. There is no globally harmonized regulatory guidance for the nonclinical safety assessment of novel excipients or guidance specific to safety testing of novel excipients in LNPs. Given the complexity of these LNP formulations, most nonclinical safety studies to support development are conducted with the drug product or with a LNP that contains non-active cargo. Three case studies (Onpattro®, Comirnaty®, and SpikeVax®) highlight that specific assessments may differ depending on the encapsulated modality, the intended use (e.g., therapeutic versus preventative vaccine), dose, and frequency of dosing. These case studies also suggest that regulatory agencies are open to scientific rationale to justify why certain tests should or should not be performed. As more products are approved, it will be important to understand how precedents set for approved products can be leveraged and what additional unique strategies may be applied to ensure nonclinical safety assessments are predictive, relevant, and meaningful for human safety. Proactive alignment with regulatory authorities will be critical in this context, especially as new approaches are proposed. Guidance documents may need to be revised or created as more experience is acquired to reflect the unique considerations for these novel excipients.
Introduction
Nucleic acid drug delivery with lipid nanoparticle (LNP) formulations has enabled novel therapeutics and vaccines to be developed (see O’Brien Laramy and Foley 2025). LNPs are used as delivery systems for a wide variety of nucleic acid cargos, including plasmid DNA (pDNA), antisense oligonucleotides (ASOs), small interfering RNA (siRNA), microRNA (miRNA), small activating or self-amplifying RNA (saRNA), messenger RNA (mRNA), and CRISPR-based gene and base-editing modalities.2-5 Accordingly, the applicable regulatory guidelines and nonclinical development strategies may vary based on the nature of the cargo, the indication, and the intended use.
LNP formulations can be composed of both naturally occurring (e.g., cholesterol) and non-natural, synthetic lipid excipients. The interaction of the nucleic acid cargo with the lipid excipients and other formulation components gives rise to significant complexities in both structure and function. The complexity of these drug delivery vehicles can complicate the safety assessment of the final drug product and its components.
The Novel Excipients Working Group (NEWG) within the International Consortium for Innovation and Quality (IQ) in Pharmaceutical Development is focused on the advancement of consensus-based scientific approaches to facilitate innovation in the development of novel excipients. A sub-team was formed in the NEWG to share current practices in the nonclinical safety assessment of novel lipid excipients contained in LNP formulations, to identify gaps in current regulatory guidance documents on this topic, and to advocate for change to facilitate the use of novel lipid excipients. The scope of the current discussion is focused on the nonclinical safety assessment of lipid excipients used in LNPs, with a focus on those considered as “novel” excipients. Here, the term “novel” indicates that the excipients are not contained in any approved drug product or approved for a specific “context of use” (e.g., dose, route of administration, duration of exposure, patient population). 6 The descriptions and recommendations provided are intended to cover nonclinical assessments to support regulatory filings from clinical development through to commercialization. Where applicable, studies needed to support specific phases of development are indicated. For considerations related to chemistry, manufacturing, and controls strategies for novel lipid excipients, readers should refer to a recent paper from the IQ NEWG 63.
Regulatory Guidance
There is no globally harmonized regulatory guidance for the nonclinical safety assessment of novel excipients, and existing guidance is limited and somewhat dated. 6 Furthermore, there are no guidance documents specific to safety testing of novel excipients in LNPs.
Regulatory Guidance Documents and Publications Related to the Safety Assessment of Novel Excipients in LNPs.
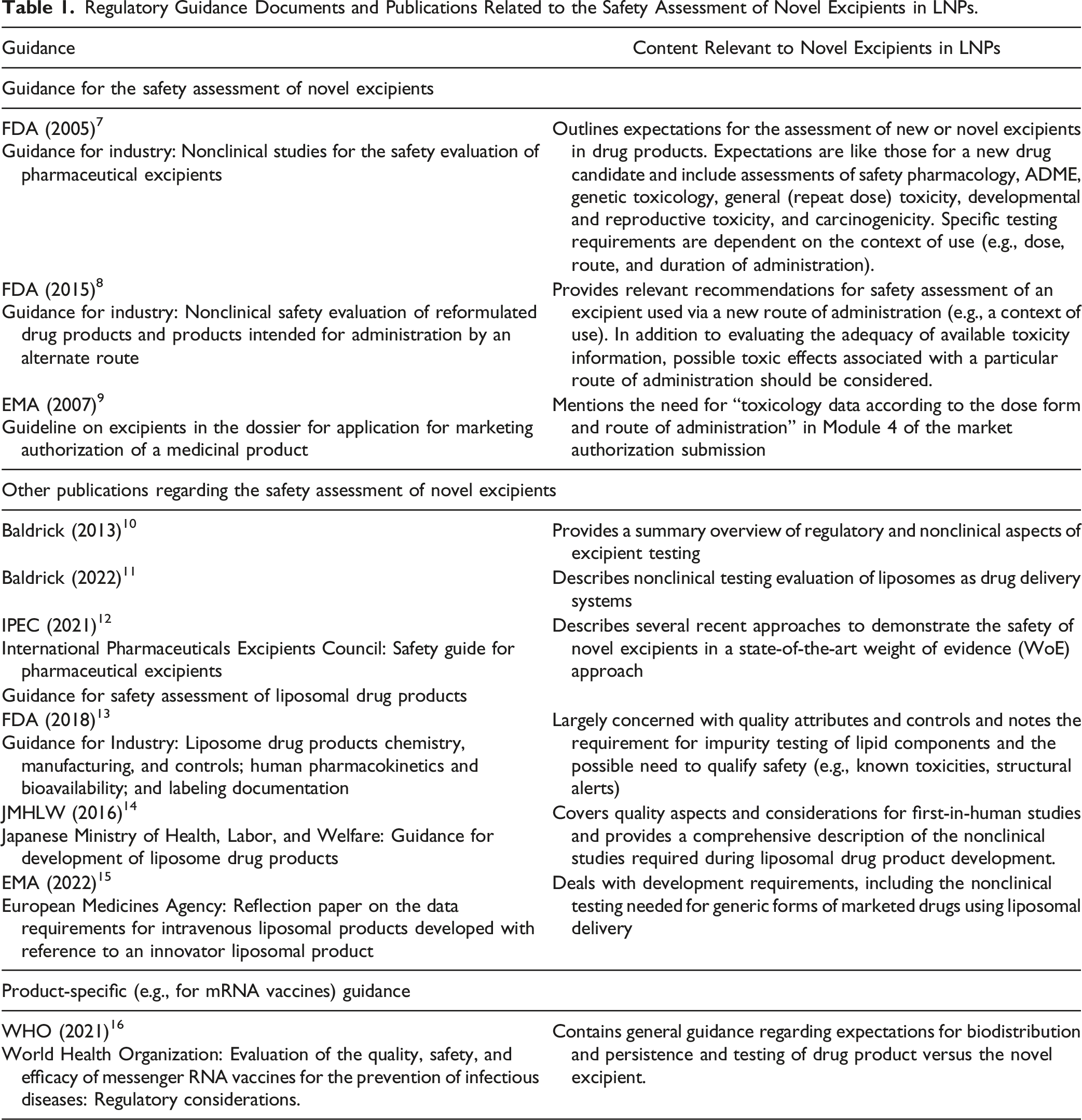
Nonclinical Testing Strategy
LNPs enhance the delivery and activity of their nucleic acid cargo by protecting the nucleic acid from nuclease metabolism, enabling cellular uptake via endocytic pathways, and promoting endosomal release of the nucleic acid cargo into the cytoplasm. 2 As such, the pharmacological, pharmacokinetic (PK), and toxicological properties of a nucleic acid delivered in an LNP are likely to differ dramatically from those of the nucleic acid administered alone. It is, therefore, scientifically justified to conduct all (or the majority of) nonclinical safety studies to support development with the drug product (active cargo within the LNP). Occasionally, it may be justifiable to use an LNP that contains a cargo other than that in the drug product which is non-active (e.g., a nucleic acid that lacks activity against the target or is not translated) to understand any sequence-specific effects of the drug product cargo or, for example, to use encoding reporter proteins to study biodistribution. However, the non-active cargo needs to be carefully considered because it may have off-target activity and thus a different toxicologic profile than the active cargo used in the drug product. This stance is consistent with recommendations made by the Oligonucleotide Safety Working Group. 19
Given the multi-functional role of the lipid excipients in the LNPs, it is worth considering how nonclinical testing of these excipients may differ from many other classes of excipients. Most conventional parenteral excipients fall into categories like buffers, tonicity modifiers, cryoprotectants, antioxidants, and surfactants whose primary function is to help achieve a target product profile, ensure stability of the drug product, or to make the product easier to administer. However, these more conventional excipients will largely dissociate from the active drug upon administration and distribute independently, and thus it is reasonable to test these excipients on their own in safety assessments.
For LNPs and many drug delivery systems, the excipients should not be tested independently in vivo. Properties like distribution and the mechanism of action derive from the LNP, not the individual lipids. Testing the individual lipids would result in differential disposition (e.g., they would not enter cells via the same pathway or may end up in different tissues) and could consequently lead to different safety outcomes that are of limited relevance to the excipient’s function in the context of an LNP. In vitro studies to evaluate the disposition and hazard (e.g., in vitro genotoxicity and drug–drug interaction studies) of the individual novel excipients in the context of the LNPs can be of value, although such testing may be challenging to perform due to solubility and bioanalytical limitations.
In general, regulatory guidance acknowledges that separate nonclinical safety studies of the novel excipients alone may (or may not) always be necessary or appropriate. For example, in Japanese Ministry of Health, Labor and Welfare (JMHLW) (2016),
14
the guidance states: Safety evaluation of the liposome components as excipients can be performed with the complete drug formulation (the whole liposome drug product) […]. However, a toxicity evaluation of the components alone may be required when a suitable toxicity evaluation derived from the liposome components cannot be performed by using only the whole liposome drug product (e.g., because of novel toxicity concern derived from the lipid structure or the potential for accumulation of lipid components).
Similarly, WHO 2021
16
states: Because the lipids used to formulate the LNPs affect the overall charge of the particle, when using LNPs made with novel lipids or when the LNPs are themselves modified (for example, altered ratios or modified processes) and these LNPs have not previously been nonclinically and clinically tested in mRNA products encapsulated in LNPs, then evaluation of the toxicity of the new formulation containing the novel lipids (or any novel excipients) may be required. Furthermore, […] genotoxicity and systemic toxicity of the novel lipid component [may be required] […] similar to the expectations for novel adjuvants set out in the WHO Guidelines on the nonclinical evaluation of vaccine adjuvants and adjuvanted vaccines (WHO 2014)
20
and/or those for new chemical entities in the ICH guideline S2 (R1).
21
Disposition
A thorough understanding of the absorption, distribution, metabolism, and excretion (ADME) characteristics of the LNP, its nucleic acid cargo, and its novel excipients is required. A biodistribution study is expected and should address whether the nucleic acid and the LNP (or lipid components) distribute away from the tissue into which the drug product was administered, into which tissues they distribute, and how long they persist. 16
Bioanalytical
Study of the individual components in in vitro systems can be challenging, in terms of solubility and protein binding. Due to these challenges, a fit-for-purpose method development and validation program for all bioanalytical assays is necessary. Fit-for-purpose is a process that allows defining the assay development and validation with the intended use of the assay. Generally, this approach leads to a continual assessment of the assay’s performance relative to its use throughout different stages of drug development with increasing levels of assay validation needed to support late-stage clinical trials. An additional consideration when determining the degree of assay validation is whether the drug product bioanalytical assay results are being used as a primary/safety versus secondary/exploratory endpoint, with a greater emphasis placed on methodological rigor for primary endpoint assays.
Pharmacokinetics and Biodistribution
Administration of LNPs is typically via intravenous or intramuscular (e.g., for vaccines) routes. Oral delivery is not currently feasible due to limited absorption and/or the instability of the active component in the gastrointestinal tract. While other local routes of administration, such as intratracheal, intrathecal, and intravitreal administration are also being investigated, such programs are less mature, with limited publicly available information. Such programs will likely require unique route-specific safety considerations. Consequently, the discussion herein is focused on the two routes with commercial precedence, namely, intravenous and intramuscular administration.
Due to rapid clearance and tissue uptake, plasma concentrations of the lipid excipients, measured in both nonclinical and clinical settings, are generally transient, and pharmacology is more closely associated with tissue concentrations. Tissue concentrations are therefore important to understand in nonclinical species, given the limited practicality to collect such data in humans and the long-lived nature of lipid excipients, which can complicate a human mass balance. 22 In these nonclinical studies, it is important to understand tissue residence times to determine durability and potential accumulation of the novel lipid excipients. The risk of tissue accumulation is particularly important for products intended for chronic administration. Issues with chronic, repeated dosing were a major concern for previous generations of permanently charged cationic lipids. 23 Current versions of LNPs incorporate transiently charged or ionizable lipids that can have a neutral charge in plasma and improved biocompatibility.
LNPs are typically taken up through endocytosis, where the environment can trigger the release of lipids and cargo into the cytosol or recycle these components to the cell surface. If recycled to the cell surface, this can result in a plasma profile that can be misinterpreted as enterohepatic recirculation. This phenomenon is most relevant to lipid excipients and oligonucleotide cargos and is not often observed in nonclinical species (although sometimes in the monkey) but is often seen in humans. 24
Dedicated biodistribution studies in nonclinical species are useful to investigate tissue distribution. Early studies of biodistribution may involve analysis of specific tissues with unlabeled material. However, it’s important to note that LNP cargo may not correlate with lipid distribution due to poor expression in certain tissue or cell types or dissociation of the lipid from the LNP. For example, polyethylene glycol (PEG) 2000—dimyristoyl glycerol (DMG) is designed to dissociate from the LNP in systemic circulation to enable protein corona formation on the LNP and enrichment in ApoE. This protein corona then enables the engagement of low density lipoprotein receptors (LDLRs) on hepatocytes and preferential distribution into the liver.25,26 For products where the LNP does not remain fully intact in circulation, the plasma and tissue pharmacokinetic profiles may not be consistent across lipids. Consequently, it is critical to directly measure the lipids in distribution studies. If lipids are difficult to track in vivo due to metabolism, degradation, or matrix interference effects, it may be helpful to use radiolabeled or fluorescently labeled lipids to confirm the biodistribution profile of lipids. Collectively, these distribution studies can help identify potential tissues or cells where toxicity concerns may arise.
In general, nonclinical PK studies are not required to support the development and licensure of a vaccine for an infectious disease indication, in agreement with WHO guidelines.27,28 Exposure to the vaccine is assessed by evaluation of immunogenicity. However, distribution studies should be conducted for new formulations or novel adjuvants or excipients to characterize their tissue distribution and persistence. 10 Agreement on the need for and design of these studies should be sought from regulatory authorities.
Metabolite Characterization
While novel lipid excipients may not be pharmacologically active, their presence, particularly in target tissues, may be of toxicologic significance. Therefore, it is important to understand the metabolism of novel lipid excipients in vitro and in the nonclinical species used in toxicology testing. In vitro testing can be performed with the novel lipids on their own, dissolved in an organic solvent, whereas any in vivo testing must be performed in the context of the intended drug product. If metabolite profiles are similar in at least one animal test species at adequate exposure levels (approximately equal to or greater than human exposure), it can be concluded that the metabolites contribution to the overall toxicity assessment has been established. 29
Drug–Drug Interactions (DDI)
Evaluation of drug interactions for LNP components can be a challenge. In vitro Drug–Drug Interaction (DDI) studies using traditional approaches for cytochrome P450 (CYP) and transporter inhibition and substrate assessment may prove difficult with lipid components due to solubility limits. Furthermore, it can be difficult to determine clinically relevant concentrations for in vitro studies. Circulating plasma concentrations, particularly at early points where concentrations are highest, are not reflective of individual lipid components available for interaction. Additionally, estimates of tissue concentrations may overestimate free concentrations due to endosomal trapping. Protein binding data may be useful to help assess clinical impact of any in vitro DDI data, but as noted above may be experimentally challenging.
One approach developed for CYP inhibition and induction studies is the use of primary human plated hepatocytes to test LNPs as opposed to individual components. This has the advantage of achieving concentrations that are clinically relevant and mechanistically similar to in vivo conditions and may be useful for discharging a potential DDI risk. A positive DDI result may require additional studies as quantitative clinical translation may be difficult.
Toxicology
As previously noted, individual novel lipid excipients are not typically tested in stand-alone safety pharmacology or toxicology studies. The study of the novel lipid excipients alone in vivo can be challenging, as many are not (or are only marginally) soluble in conventional vehicles for safe administration to animals. Instead, these excipients are tested within the context of the drug product. Similarly, studies of local tolerance are typically performed with the drug product and not the individual novel excipients. Developmental and reproductive toxicology studies are also conducted within the context of the drug product and not with individual novel excipients. Assessment of the genotoxic risk for individual novel excipients may be considered in the development strategy. The potential effects of LNPs and lipid excipients on the immune system are a known concern and should be considered in the development strategy.
Genotoxicity Studies
For small molecule drug substances, two in vitro and one in vivo genotoxicity assays are required. 30 As small molecules, ICH S2 (R1) guidance would apply to lipids used in LNP formulations. As described above, the in vitro assays may be conducted with each novel excipient (although the concentration range may be limited by solubility concerns), while in vivo assays are typically conducted with the drug product.
As indicated in the WHO guideline, 16 genotoxicity of the novel lipid excipients in vaccines should be assessed, similar to the expectations for novel adjuvants set out in the WHO Guidelines on the nonclinical evaluation of vaccine adjuvants and adjuvanted vaccines 28 and/or those for new chemical entities in the ICH guideline S2 (R1). 21 Alternatively, the Sponsor may issue a paper justification for no or low risk associated with novel excipients. The lack of genotoxicity concern may be driven by the absence of structural alerts for mutagenicity (e.g., from in silico analyses) and the fact that exposure to the novel excipients is low. For vaccines, in vivo genotoxicity studies are not required.
Developmental and Reproductive Toxicology
When warranted, developmental and reproductive toxicology (DART) studies are conducted with the drug product. The plasma kinetics of the nucleic acid cargo and the novel excipients should be considered when designing the dosing regimen in these studies to ensure sufficient maternal exposure during the key reproductive and developmental milestones. 31 For example, daily dosing during the period of organogenesis is the standard design in rat and rabbit embryo-fetal development studies; however, these daily dosing regimens with LNPs may lead to plasma and tissue drug accumulation of novel lipids and associated maternal toxicity. Less-frequent dosing schedules can be employed for LNPs if the novel lipids reside in the maternal circulation for longer than 1 day with the dosing schedule during organogenesis based upon the respective plasma half-lives of the novel lipids. If prolonged maternal exposure is lacking, smaller daily doses during organogenesis (based on fractionating the weekly or monthly doses used in general toxicity studies to their daily equivalents) can be administered. Another possible design could be, for example, to dose different cohorts of dams on different gestational days. However, caveats of this type of design center around the 3R’s of animal use and this type of design is generally not advisable. In all cases, the aim is to provide an adequate AUC-based safety margin (based on the cargo), unless limited by maternal toxicity, to the proposed clinical dose after normalizing the AUC to the clinical dosing frequency.
Determination of a pharmaceutical’s concentration in the embryo or fetus or in milk, may be useful to interpret discordant or equivocal evidence of a potential developmental hazard. However, results from these nonclinical hazard evaluations must not be used to directly project human exposure. Before conducting such evaluations, a weight-of-evidence approach for determining the likelihood of fetal or lactational exposure to the LNP or its constituents should be employed. For example, properties of novel excipients such as lipophilicity and molecular weight will influence exposures. Development and validation of bioanalytical methods for these matrices should also be considered for GLP studies.
Effects on the Immune System
LNPs and their nucleic acid cargo are known to potentially affect the innate immune system in a variety of ways.
32
As stated in WHO (2021)
16
: Both the mRNA molecules and the LNPs (which enable successful delivery and cellular uptake) have properties that can influence and trigger the innate immune system […]. Other components added to aid delivery, such as PEG, although relatively benign, can also influence the physicochemical properties and thus the safety profile [...]. It is therefore important to understand the overall product profile including the formulation and how physicochemical properties (which may vary) can influence inflammation and the safety profile.
LNPs and their constituents can be detected by cell surface or intracellular innate immune receptors, 33 and the combination or synergistic activation by both the LNP and nucleic acid may lead to the secretion of pro-inflammatory cytokines. This response could be favorable in the context of the local administration of a vaccine 34 or could lead to tissue toxicity and adverse event outcomes in the context of systemic exposure with a therapeutic. 35
Immune responses are dependent on the composition (e.g., the specific lipid excipients used), the RNA cargo (e.g., the design of the cargo), the presence of impurities, the LNP properties (e.g., size, surface charge), the route of administration, the biodistribution profile of the LNPs, and whether patients are pre-treated with anti-inflammatory medications.36,37 These differences can enable very different dose levels to be administered to humans, as seen with Onpattro® relative to Spikevax® or Comirnaty®. At present, the community does not fully understand how to systematically tune immune activation for specific use cases, nor how responses observed in nonclinical models will translate into humans.
For this reason, novel excipients and cargos are usually tested as part of the LNP in accordance with ICH S8. 38 However, ICH S8 does not provide detailed guidance for immune safety testing of LNPs or novel lipid excipients. In recent years, several white papers describing the evaluation of nonclinical immune safety have been published19,39–41 and serve as helpful guides.
Pro-Inflammatory Cytokines
LNPs can interact with specific pattern recognition receptors (PRR), like Toll-like receptors (TLRs), that can result in adverse effects that may limit dosing or diminish the therapeutic effect.32–34 Such adverse responses include cytokine release syndrome (CRS) associated with pro-inflammatory cytokines and/or lymphocyte activation. In humans, dose-dependent cytokine release can result in adverse events that are generally mild-to-moderate and consist of fever, nausea, chills/rigors, tachycardia, headache, rash, dyspnea, and in some cases, transient hypotension with effects occurring 4–8 hours following drug infusion.44,45
In vitro systems, in particular those employing human whole blood, peripheral blood mononuclear cells (PBMCs) and serum/plasma, can be incorporated in preliminary hazard evaluation screens to assess cytokine release, complement activation, and immune cell profiling.46–49 These systems can be translatable but are subject to donor-to-donor variability and require testing multiple donors. Monitoring cytokine induction in in vivo nonclinical toxicology species is generally recommended for LNP-based therapeutics. 19 Cytokine and chemokine evaluation should include but may not be limited to IL-1 β, IP-10, IL-6, IL-8, TNF-a, MCP-1, and IFN-g. Cytokine levels typically can be observed systemically within 6 hours in serum of non-human primates following IV administration and generally return to baseline within 24 hours post-dose. The route of administration can also affect the pharmacokinetics of cytokine production.
However, the translation of nonclinical models used to evaluate innate immune system responses to humans is often poor, and evaluation in primary human cells or whole blood is important. It is therefore useful during species selection for toxicology studies to understand the human relevance of the immune response to LNPs and novel excipients in rodent and non-rodent species. Rodents tend to have a different expression pattern and ligand specificity of PRRs, as well as tolerance to cytokine release compared to humans.47,50 Interestingly, Tahtinen et al. (2022) 47 demonstrated that lipoplex or LNP mRNA vaccines induced innate immune responses driven by a robust upregulation of IL-1ra in mice. This upregulation rendered the mice much more tolerant to vaccine-induced, IL-1-mediated cytokine release compared to humans. Conversely, IL-1ra knockout mice had an increased sensitivity to cytokine release, with a preferential IL-1β response more similar to humans. The authors also showed that nonhuman primate (NHP) responses to these mRNA vaccines were more similar to mice than humans when it came to IL-1ra induction levels in vitro. These observations have been further supported by in vivo findings, as different LNPs have been shown to induce transient elevation in serum IL-1ra in NHPs.51,52 In fact, the potential for these types of effects is sometimes managed clinically by administration of a steroid prior to administration of the LNP-containing drug product.
Cytokines can also be assessed in rodent species for LNP-based vaccines. These assessments are useful from a mechanistic perspective, but care must be taken to ensure that appropriate assays are in place, including careful attention to the matrix, timepoints, and controls. Some cytokines of importance, such as IL-1β, are typically more locally acting. Consequently, while these locally acting cytokines may be of interest in the context of vaccines, assessment may not be plausible unless a robust assay can be developed. Measurement of other cytokines as indicators of IL-1 signaling, such as IL-6 or IL-8, can also be utilized.
Complement Activation
LNPs and other nanomedicines have been shown to interact with various plasma proteins including complement factors.26,53 This has been linked to hypersensitivity reactions (HSRs) such as complement activation-related pseudoallergy (CARPA).1,54 Complement activation is routinely assessed in nonclinical models, such as rats and non-human primates, by quantifying cleaved products (e.g., C3a, C5a, Bb, and C5b-9) by ELISA directly in serum or plasma following administration.40,54,55 However, results of complement assays from animal models do not always predict clinical reactions. Consequently, it is important to assess complement status at multiple timepoints (e.g., 2–6 hours post-dosing), as it can contribute to mechanistic insights into HSRs and aid in human risk assessment. Recently, the use of a hypersensitive animal model, such as pigs in the context of Comirnaty®, has shown that CARPA may help explain rare HSRs observed in the clinic.56,57 However, further validation is needed to understand the relevance of LNPs reactions in pigs to the response observed in humans and the contribution of other non-complement mediated mechanism(s) for these reactions. The use of such sensitive models, along with assessment in human in vitro systems (e.g., whole human blood), may help to bridge the gap in translatability of HSRs. Current approaches to reduce the intensity of HSRs include premedication with immunosuppressive drugs (e.g., corticosteroids) and slow infusion rate which are now adapted in clinical protocols resulting in low HSRs in most patients. 58 While such measures can reduce HSRs, it remains a challenge to predict or prevent these reactions considering their complex pathophysiology.
Immunogenicity
Nonclinical species and humans can develop anti-drug antibodies (ADA) against LNPs following systemic exposure. This response is largely thought to be elicited by the polyethylene glycol (PEG)-containing lipid; however, the magnitude of responses can be tuned to enhance or diminish immune responses based on the LNP composition. Modifications that could reduce immunogenicity include: lipid composition, which can affect the stability and uptake and immune cell interaction; size, as larger particles can more easily be recognized by the immune systems and provoke a response; surface charge, which can modulate the type and magnitude of immune response; and pH-sensitivity, which can enhance antigen processing and presentation. 59
Immunogenicity (formation of anti-PEG antibodies) to pegylated drugs has been widely recognized and in some cases, can lead to altered tissue biodistribution and accelerated blood clearance of these pegylated compounds (as reviewed by Chen 2021). 60 ADA titers are therefore measured in repeat-dose toxicology studies (and in clinical studies) to aid in the interpretation of drug exposure and effects. Baseline assessments of ADA levels are recommended, as naïve animals and human subjects may have pre-existing anti-PEG antibodies which may (or may not) neutralize the LNP drug product. Importantly, immunogenicity to biotherapeutic drugs in nonclinical species may not be predictive of potential immunogenicity in humans.61,62 Therefore, the ADA results in animals may be of limited value in the prediction of responses in humans.
Qualification of Impurities
Residual impurities in pharmaceutical products can arise from starting materials, reagents, manufacture, or from degradation during storage and in-use conditions. Some impurities may pose a potential health risk to humans, and thus a risk assessment of organic, inorganic, and potential mutagenic impurities is essential during drug development. It is important to consider impurities related to novel lipid excipients during an impurity risk assessment of LNP drug products. Risk assessment includes both CMC aspects (e.g., impurity characterization, reporting, specification limits, and control strategies) 1 as well as safety aspects (impurity qualification and assessment) of impurities present in lipid excipients.
ICH quality guidelines provide recommendations for impurity threshold and qualifications based on amount of impurity in relevant safety studies to that present in new drug substance 63 or drug product. 64 Assessment of potential genotoxic impurities, including nitrosamines, is described in the ICH M7R2 guidance. 65 The latter two guidance documents do not explicitly reference LNPs but may be applied to this context for the assessment of novel excipient impurities. 65
The need to evaluate extractable and leachable impurities for the drug product is outlined in FDA Guidance.66,67 These studies should be performed with the drug product.
Case Studies
Studies With Novel Excipients in LNPs (From Case Studies).

ONPATTRO®
ONPATTRO® (patisiran), approved in 2018, is an LNP formulation of siRNA for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults.13,71 The product is administered IV every 3 weeks at an siRNA dose of 0.3 mg/kg. ONPATTRO® contains two novel, non-compendial lipids, DLin-MC3-DMA and PEG2000-C-DMG, and two non-novel structural lipids, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and cholesterol.
Disposition
The pharmacokinetics of the oligonucleotide cargo and both individual novel lipids were fully characterized. It was important to evaluate the ADME properties of each of these components in vitro and in vivo. These evaluations necessitated a comprehensive method development and validation program for all the bioanalytical assays used to measure the oligonucleotide and the two novel lipids (and metabolites) in the nonclinical development program.
The pharmacokinetic parameters of the siRNA, DLin-MC3-DMA and PEG2000-C-DMG were examined in mouse, rat, and monkey. Distribution to various tissues (most notably liver and spleen) as well as excretion was investigated using unlabeled drug product and an LNP formulation containing radiolabeled 14C-DLin-MC3-DMA in rat and monkey. In vitro metabolic stability of siRNA, DLin-MC3-DMA and PEG2000-C-DMG was examined in serum and liver S9 in rodent and human. Metabolite profiles in various matrices from in vivo samples from rat, monkey and human were evaluated. In vitro studies for CYP enzyme inhibition, induction and transporter interactions were investigated for the siRNA, DLin-MC3-DMA, and PEG2000-C-DMG.
The two novel excipients (DLin-MC3-DMA and PEG2000-C-DMG) along with the oligonucleotide cargo were measured in fetuses of rats and rabbits after maternal exposure. Exposure to these excipients was also measured in rat milk.
Safety Pharmacology, Pharmacodynamics, and Toxicology
Neither novel lipid was assessed individually in safety pharmacology or toxicity studies. Instead, the nonclinical studies were conducted with the drug product.
Genotoxicity
The two novel excipients were tested in in vitro genotoxicity assays (i.e., bacterial mutagenesis and chromosome aberrations). An in vivo mouse bone marrow micronucleus study was also conducted using the drug product.
Challenges arose when conducting the in vitro assays with the two novel lipids due to their limited solubility in the standard solvents utilized in these assays. 30 Preliminary solubility assessments were necessary prior to designing the pivotal genotoxicity assays, and the concentration range evaluated was limited by these solubility concerns.
Developmental and Reproductive Toxicology (DART)
Rodent and rabbit DART studies were conducted using the drug product. In addition, an LNP bearing a rodent-active pharmacological active surrogate oligonucleotide was also administered to evaluate potential effects due to primary and secondary pharmacology.
Carcinogenicity
Carcinogenicity of the drug product was assessed in a 6-month bioassay with TgRasH2 mice. A waiver was obtained for the 2-year rat carcinogenicity study because of immunogenicity-limited exposure in the chronic repeat-dose rat toxicology study.
Immunotoxicity/Immunogenicity
Administration to monkeys resulted in transient elevations in systemic cytokines (IL-1ra and IL-6) as well as increases in complement split products (C3a or Bb) at 15 minutes to 24 hours after dosing. These changes were not accompanied by any adverse clinical signs.
Titers of IgM and IgG directed against the PEG2000-C-DMG lipid were determined in early repeat-dose toxicology studies. Subsequent validated combined (IgM and IgG) methods were used to measure ADA in the chronic toxicology studies in rats and monkeys. ADA against PEG2000-C-DMG were present in both species but at low incidence, and titer in monkeys were without effect on pharmacodynamics or exposure. In contrast, a robust ADA response to PEG2000-C-DMG was observed in the chronic rat toxicology study with ONPATTRO®, and resulted in altered exposure to the siRNA, DLin-MC3-DMA, and PEG2000-C-DMG.
Impurities
For both novel excipients toxicological qualifications of potential organic impurities in final drug product were performed using the No-Observed-Adverse-Effect-Levels (NOAEL) from relevant nonclinical studies. NOAELs for the LNP were based on the siRNA component of the LNP. To determine the qualification level of lipid impurities, the dose levels of each novel lipid at the NOAEL were determined. Then, the amount of each lipid impurity at the lipid’s dose level in the toxicology batch(s) was quantified and used to qualify comparable levels in the clinical batches of drug product.
For genotoxic impurity assessment, reactive structural alerts in the synthesis pathway of each novel excipient were identified and evaluated for mutagenicity and carcinogenicity potential using database searches and/or in silico methodologies (DEREK and Leadscope®), and an acceptable intake per dose was determined based on either the threshold of toxicological concern (TTC) or the permissible daily exposure (PDE) value. The acceptable level of each impurity in the final drug product was determined by dividing the acceptable intake per dose by the estimated human dose of the novel lipid excipient. A determination was then made as to whether each impurity would likely be present in the final drug product at amounts that exceeded the acceptable human levels by evaluating purification processes and/or control strategies before assigning an ICH M7 Classification Level to each.
No additional in vivo toxicology studies for organic impurities or bacterial mutagenicity assays for any of the putative genotoxic impurities were required.
As part of the Japanese marketing application for ONPATTRO® a literature-based toxicological assessment of all excipients in the LNP, including the two novel ones and the two structural lipids, DSPC and cholesterol, was included. This assessment provided published information on the biological roles of these lipids (or structurally similar naturally occurring lipids) and doses of the structural lipids in ONPATTRO® compared to those of other approved liposomal drugs that contained these lipids.
COMIRNATY®
COMIRNATY® (tozinameran), 68 a COVID-19 mRNA (BNT162b2) vaccine formulated with LNPs, is indicated for the prevention of coronavirus disease 2019 (COVID-19) in humans.69,72 The vaccine is given as a 30 μg dose of mRNA via intramuscular administration. COMIRNATY® contains four lipid components: cholesterol is a compendial excipient and the other three lipids, ALC-0159, ALC-0315, and DSPC, are non-compendial excipients.
The functional lipid excipients ALC-0315 and ALC-0159, were classified as novel excipients. Both structural lipids DSPC and cholesterol are used in several already approved finished products. A justification was provided for why DSPC is not considered a novel excipient. This justification argued that DSPC is used as part of the LNP for the EU approved drug product ONPATTRO®, which is administered IV in a much higher dose than the IM dose for COMIRNATY®. Additionally, 1,2-Dioleoyl-sn-Glycero-3-Phosphocholine (DOPC), a structurally related lipid, is present in EU-approved drug products for IM administration. The level of information provided for DSPC was considered sufficient and appropriately justified, in line with the requirements for a known excipient.
Disposition
The PK, biodistribution, and metabolism of COMIRNATY® and the two novel lipids (ALC-0315 and ALC-0159) contained in the formulation were studied via in vitro systems and in vivo.
The biodistribution of the two novel lipids was assessed in plasma and liver after IV administration of an LNP-formulated luciferase RNA in rats. In addition, biodistribution of luciferase was assessed in mice after the IM administration of an LNP-formulated luciferase RNA via in vivo bioluminescence. Last, the biodistribution of a [3H]-labeled luciferase mRNA LNP formulation was studied in rats after IM administration.
Results indicated that metabolism of ALC-0315 and ALC-0159 appears to occur slowly in vitro and in vivo. ALC-0315 and ALC-0159 are metabolized by hydrolytic metabolism of the ester and amide functionalities, respectively, and this hydrolytic metabolism is observed across the species evaluated.
Safety Pharmacology, Pharmacodynamics, and Toxicology
No secondary pharmacodynamic, safety pharmacology or pharmacodynamic drug interaction studies were conducted with COMIRNATY® due to the nature of the RNA-based vaccine product (consistent with WHO 2005). 27
The toxicity of COMIRNATY®, and other vaccine candidates, was studied in two repeat-dose toxicity studies in rats. As the full human dose was used in rats, there was a large dose differential between the human COMIRNATY® dose, and the dose used in the toxicity studies provided an acceptable safety margin.
Nonclinical GLP-compliant studies in Wistar Han rats were conducted to assess the local tolerance, systemic toxicity, and immune response to four modified-nucleoside RNA vaccine candidates encoding immunogens derived from the spike (S) glycoprotein of SARS-CoV-2, encapsulated in LNPs. 73
The complete formulation (modified RNA within LNPs) was assessed in toxicology studies. No toxicological assessment was performed on the LNP alone or its specific novel excipients. The novel LNP components were not considered primarily as adjuvant substances.
Genotoxicity
No genotoxicity (or carcinogenicity) studies were submitted. The components of the vaccine formulation are lipids and RNA that are not expected to have genotoxic potential.
The novel excipient ALC-0159 contains a potential acetamide moiety. Based on literature data, acetamide genotoxicity is associated with high doses and chronic administration (≥1000 mg/kg/day). Since the amount of ALC-0159 excipient in the finished product is low (50 μg/dose), its clearance is high and only two administrations of the product are recommended for humans, the genotoxicity risk is expected to be very low.
Developmental and Reproductive Toxicology
A fertility-embryofetal development study in rats was conducted with COMIRNATY® after IM administration. 75
Carcinogenicity
No carcinogenicity studies were reported, in line with WHO guidance27,28 for vaccine development which states these studies are not required for vaccine antigens.
Immunotoxicity/Immunogenicity
Activation of the innate immune system following IM administration of an LNP-formulated luciferase reporter RNA was characterized in mice. Cytokine analysis was also included in one of the repeat-dose toxicity studies with BNT162b2 in rats after IM administration.
Impurities
Lipid-related impurities were identified in the finished drug product and were characterized. Based on the low dose (30 μg mRNA), the amounts of lipid related impurities observed in produced finished product batches were considered too low to be of toxicological significance.
SPIKEVAX®
SpikeVax® (mRNA-1273) is an mRNA-LNP vaccine against SARS-CoV-2, indicated for active immunization to prevent COVID-1913.74,75 The vaccine is given as a 50 μg dose of mRNA via intramuscular administration. The modified mRNA in SpikeVax® encodes for the full-length S protein of SARS-CoV-2 and is modified to introduce two proline residues to stabilize the S protein in a prefusion conformation (S-2P). The modified mRNA is encapsulated in an LNP that contains four lipids: SM-102 (a custom-manufactured, ionizable lipid), PEG2000-C-DMG, cholesterol, and DSPC. 76
In the initial US and EU filings, Moderna proposed that SM-102 and PEG2000-C-DMG were not novel excipients, but rather were starting materials in their drug substance. 77 This argument was accepted by regulators in the US. By contrast, regulators in the EU pushed back against this classification, and designated both lipids as novel excipients, in line with ONPATTRO® and COMIRNATY®.
Disposition
No dedicated ADME studies with SpikeVax® were conducted.
The evaluation of SpikeVax® tissue distribution was based on a platform approach. A rat biodistribution study using a similar mRNA-based vaccine encoding cytomegalovirus antigens (mRNA-1647) was used to support biodistribution of mRNA-1273. Following a single IM injection at 100 μg mRNA-1647, the plasma and tissue pharmacokinetics and tissue distribution were assessed in blood and a pre-specified set of organs/tissues over a 120-hour period. A qualified branched DNA (bDNA) multiplex method was used.
Specific metabolism studies with SpikeVax® variant vaccines were conducted. Specifically, the metabolism and elimination of the amino-lipid component SM-102 in mRNA-1273 was examined in vitro and in vivo.
Safety Pharmacology and Pharmacodynamics
No studies on the secondary pharmacodynamics, safety pharmacology, and pharmacodynamics drug interactions were performed, in accordance with applicable guidelines for vaccines.
Genotoxicity
SpikeVax® mRNA contains natural nucleosides and LNPs. The genotoxic potential of the novel excipient SM-102, as well as the final vaccine formulation, was evaluated. The other lipid components contained in the final formulation (i.e., PEG2000-DMG, DSPC, and cholesterol) were not separately tested but are present in the formulation tested in the in vivo genotoxicity studies. As already mentioned, DSPC and cholesterol do not raise any concern in terms of genotoxic potential.
SM-102 was tested for its genotoxic potential in a bacterial reverse mutation test in Salmonella typhimurium and Escherichia coli and in an in vitro mammalian cell micronucleus test in human peripheral blood lymphocytes. In vivo genotoxicity testing was performed with Nascent Peptide Imaging (NPI) luciferase mRNA in SM-102-containing LNPs and an in vivo micronucleus study in rat was performed with mRNA-1706 in SM-102-containing LNPs using IV administration.
Toxicology Studies
The nonclinical safety strategy was also based on a platform technology approach. As such, GLP repeat-dose toxicity studies were performed with other LNP-mRNA products containing the same ionizable lipid (SM-102). Later, following inclusion of the variant spike, a GLP repeated dose toxicity study was performed with SpikeVax® in rats. No toxicological assessment was performed on the LNP alone or its specific novel excipients.
No stand-alone local tolerance studies were conducted, in line with relevant guidance on nonclinical vaccine development; local tolerance was evaluated in repeated dose toxicity studies.
Developmental and Reproductive Toxicology
A GLP-compliant reproductive and developmental toxicology study with SpikeVax® was conducted in female Sprague Dawley CD rats.
Carcinogenicity
No carcinogenicity studies were submitted. This is scientifically acceptable and in line with relevant guidelines on nonclinical development of vaccine candidates. The components of the vaccine formulation are lipids and natural nucleosides that are not expected to have carcinogenic potential.
Immunotoxicity/Immunogenicity
The nonclinical immunogenicity and protective activity of SpikeVax® was evaluated in young and aged mice, Syrian golden hamsters, and NHPs. Activation of the innate immune system following IM injection of an LNP-formulated luciferase reporter RNA into mice was performed with assessment of chemokines and cytokines.
To assess the potential risk for Vaccine-induced Enhancement of Disease (VAED), the type of T helper responses (Th1 vs Th2) was evaluated in mRNA-1273 immunized mice and NHPs, based on IgG subclasses measured by ELISA (IgG2a, IgG1, IgG2a/IgG1, young and aged Balb/c mice) and/or T-cell cytokines (e.g., IFN-g, IL-2, TNF-a, IL-4, IL-5, IL-10, and IL-13) measured by intracellular cytokine straining (ICS) and FACS (in young Balb/c mice, in NHPs).
In addition, cytokine analysis (usually different interleukins, interferon gamma, acute-phase proteins) was generally assessed in non-product-specific (but LNP-specific) repeated dose toxicity studies.
Impurities
A specification of impurities for the SM-102 lipid and a risk assessment for the presence of benzene in SM-102 was required by regulators.
Conclusion
The nonclinical assessment of novel lipid excipients used in LNPs requires unique considerations and approaches compared to conventional novel excipients. For example, many assessments are performed with the drug product, or a surrogate LNP, rather than the novel lipid on its own. At present, there are no unified, harmonized regulatory guidance documents to inform what assessments are required. Consequently, sponsors are left to determine what assessments are needed for their specific product and to provide adequate scientific justifications to support those strategies.
The case studies above highlight that specific assessments may differ depending on the encapsulated modality, the intended use (e.g., therapeutic versus preventative vaccine), dose, and frequency of dosing. These case studies also show that regulatory agencies are open to scientific arguments to justify why certain tests should or should not be performed. Indeed, the publicly available information about the nonclinical testing strategies employed across the case studies shows that there is not a one-size fits all strategy.
Further clarity and consistency are requested from the regulators as to whether novel lipids are classified as novel excipients. As described above, the SpikeVax® regulatory submissions argued that the novel lipids should be considered starting materials in the drug substance. The distinction between drug substance starting material and novel excipient has important consequences for regulatory review and product development, 77 including expectations for nonclinical assessments. Such distinctions may also have implications for future products that attempt to use novel lipids in commercial products. For example, if a novel lipid is classified as part of the drug substance and used as an excipient in a subsequent product, can the safety data from the prior product be leveraged (assuming appropriate permissions are in place)? Further clarity from regulators would be helpful.
As more products are approved, it will be important to understand how precedent set from approved products can be leveraged, whether regulators encourage more consistency to align with precedent, or if additional unique strategies may be employed. Furthermore, as sponsors branch into new uses of LNPs, including novel routes of administration, novel modalities, and novel indications, the nonclinical safety assessments will evolve and require unique scientific justifications. With the growing experience with such nonclinical safety approaches, researchers should continue to explore which assessments are most predictive, relevant, and meaningful for human safety. Proactive alignment with regulatory authorities will be critical in this context, especially as new approaches are proposed, and as data are generated with new models and assays. Acknowledging the challenges associated with the development and global harmonization of regulatory guidance documents that cover the variety of unique contexts in which LNPs may be used, we advocate for revisions of existing guidance documents or the development of new guidance documents as more experience is acquired and to reflect these unique considerations for novel excipients.
Footnotes
Acknowledgments
This perspective was developed under the auspices of the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ), a not-for-profit organization of pharmaceutical and biotechnology companies with a mission of advancing science and technology to augment the capability of member companies to develop transformational solutions that benefit patients, regulators and the broader research and development community. The work was sponsored by the IQ novel excipient working group (NEWG) which is a member of the Drug Product Leadership Group (DPLG). The mission of the respective groups is to improve awareness of the importance of novel excipients in facilitating new therapeutic concepts into products for patients (NEWG) and to influence the strategic direction of drug product development and manufacturing for the benefit of industry and patients (DPLG). The manuscript was reviewed by both groups and members of IQ’s DruSafe Leadership Group, whose mission is to advance nonclinical safety sciences and impact the global regulatory environment. The authors would also like to thank David Foley (Merck & Co., Inc., Rahway, NJ USA) for his role in conceptualization of the IQ CMC and nonclinical perspectives on development of LNP-containing drugs and the following individuals who reviewed the manuscript for their helpful feedback on the article: Zhao Yu and Nicholas Buss (Eli Lilly & Co., Inc.), Mayur Mitra (Genentech, Inc.), and Siri Tahtinen (Genentech, Inc.). The authors thankfully acknowledge Christine Smith (Alynlam Pharmaceuticals, Inc.) for significant formatting refinements to the manuscript.
Author Contributions
Buckley, L.A., and Sutherland, J., contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Borude, P., contributed to design, contributed to acquisition and analysis, and drafted manuscript; Broudic, K., contributed to design, contributed to acquisition and interpretation, drafted manuscript, and critically revised manuscript; Collin, P., contributed to design, contributed to acquisition, analysis, and interpretation, and drafted manuscript; Hillegas, A., contributed to design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; MacLauchlin, C., contributed to design, contributed to acquisition and analysis, drafted manuscript, and critically revised manuscript; Saleh, A., and Thomas, J., contributed to design, contributed to acquisition and analysis, and drafted manuscript; O’Brien Laramy, M., contributed to conception and design, contributed to acquisition, analysis, and interpretation and drafted manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Lorrene Buckley is an employee of Eli Lilly & Co., Inc. and may hold shares and/or stakes in the company. Jessica Sutherland, Chris MacLauchlin, and Prachi Borude are employees of Alny lam Pharmaceuticals, Inc. and may hold shares and/or stakes in the company. Philippe Collin and Amer Saleh are employees of AstraZeneca and may hold shares and/or stakes in the company. Karine Broudic is a Sanofi employee and may hold shares and/or stakes in the company. Aimee Hillegas is an employee of GSK and may hold shares and/or stakes in the company. Amy Sharma is a Pfizer, Inc. employee and may hold shares and/or stakes in the company. Justina Thomas is an employee of Merck Sharpe & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA and may hold shares and/or stakes in the company. Matthew O’Brien Laramy is an employee of Genentech, Inc. and may hold shares and/or stakes in the company.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.