
Abstract
Ventricular noncompaction cardiomyopathy is a rare form of congenital cardiomyopathy with increasing evidence of genetic etiology, especially when presenting in childhood. Fetal presentation is rare. We describe a case of fetal hydrops, presenting at 24 weeks gestation and leading to intrapartum death at 26 weeks gestation. Autopsy examination revealed characteristic features of left ventricular noncompaction. A genetic analysis identified a constellation of variants of unknown significance in MYH6, TNNC1, and MYBPC3, genes known to be important in sarcomeric function. Additionally, the variant in MYBPC3 was homozygous. While this case did not demonstrate a conventional single-gene mutation as the cause of the ventricular noncompaction, a broader genomic investigation revealed several variants in sarcomeric genes which may act synergistically to impact cardiac function.
Introduction
Ventricular noncompaction (VNC) is a presumed congenital cardiomyopathy characterized by excessive trabeculation within the ventricular wall and subsequent variable compromise of ventricular function. VNC can be isolated, or associated with other cardiac anomalies and can be asymptomatic or present at any age, usually with cardiac failure, arrhythmias, or thromboembolic disease. The etiology is under dispute and appears multifactorial. Classically, it has been suggested that VNC is a developmental disorder. The primitive heart is avascular and relies on myocardial trabeculation for direct oxygen transfer to the outer compacted layer of myocardium. As the compacted layer thickens, between the 5th and 8th week of embryonic life, the developing coronary artery circulation begins to provide oxygen to the myocardium. 1 It has been suggested that VNC is due to a maturation arrest, with likely ongoing proliferation of the trabecular layer. Alternative theories of VNC are that it arises due to an impairment of cardiac myocyte function or due to cardiac neuropathy. 2 Latterly, it has been shown that excessive left ventricular trabeculations can be detected in adults in a significant proportion of the healthy adult population, 3 leading to the assumption that excessive trabeculation, consistent with a diagnosis of VNC, may be insufficient of itself for a diagnosis of cardiomyopathy. However, the diagnosis of VNC, with appropriate cardiac symptomatology, has increasingly been attributed to a genetic etiology, with several diverse genetic mutations described in association with VNC and resulting in its classification as a primary genetic cardiomyopathy by the American Heart Association. 4 These mutations occur in genes encoding sarcomeric proteins, as well as those encoding proteins with functions as diverse as inner mitochondrial membrane enzyme activity, Notch signaling, and cytoskeletal function. 5
We describe a diagnostic, panel-based genetic analysis of a case of isolated VNC presenting as fetal hydrops at 24 weeks gestation and culminating in intra-partum death at 26 weeks.
Case Report
The mother was a 33-year-old woman with two previous miscarriages. This pregnancy was conceived after ovulation induction. She had a low risk combined first trimester fetal screen and a normal 18-week morphology scan. The mother presented at 24 weeks gestation with a history of possible ruptured membranes. This was not confirmed by clinical examination and transabdominal ultrasound scan showed normal liquor volume. However, the fetus was noted to have gross ascites and a possible pericardial effusion and was referred for a tertiary evaluation.
Tertiary scan revealed cardiomegaly (Figure 1(A)). There was severe biventricular cardiac dysfunction with almost no contractility and minimal cardiac motion. Ventricular systolic function was impaired (Figure 1(B)), and there was reversal of A wave on Doppler insonation of the Ductus Venosus, confirming severe cardiac dysfunction (Figure 1(C)). Middle cerebral artery peak systolic blood flow was normal indicating that there was no fetal anemia. The fetal heart rate and rhythm were normal. Small pericardial effusions were noted. There was a large volume of ascites with an abdominal circumference of >95th percentile for gestational age (Figure 1(D)).
Ultrasound images from obstetric scan of the fetus at 24 weeks with systolic dysfunction and fetal hydrops. A, Cardiomegaly with a globular heart filling most of the chest cavity. B, Systolic dysfunction indicated by poor excursions of the ventricular wall (solid arrows) on M mode insonation. C, Reversed A wave on Doppler insonation of the Ductus Venosus indicating cardiac failure. D, Ascites (arrow) in the fetal abdomen with polyhydramnios indicative of fetal hydrops from cardiac dysfunction.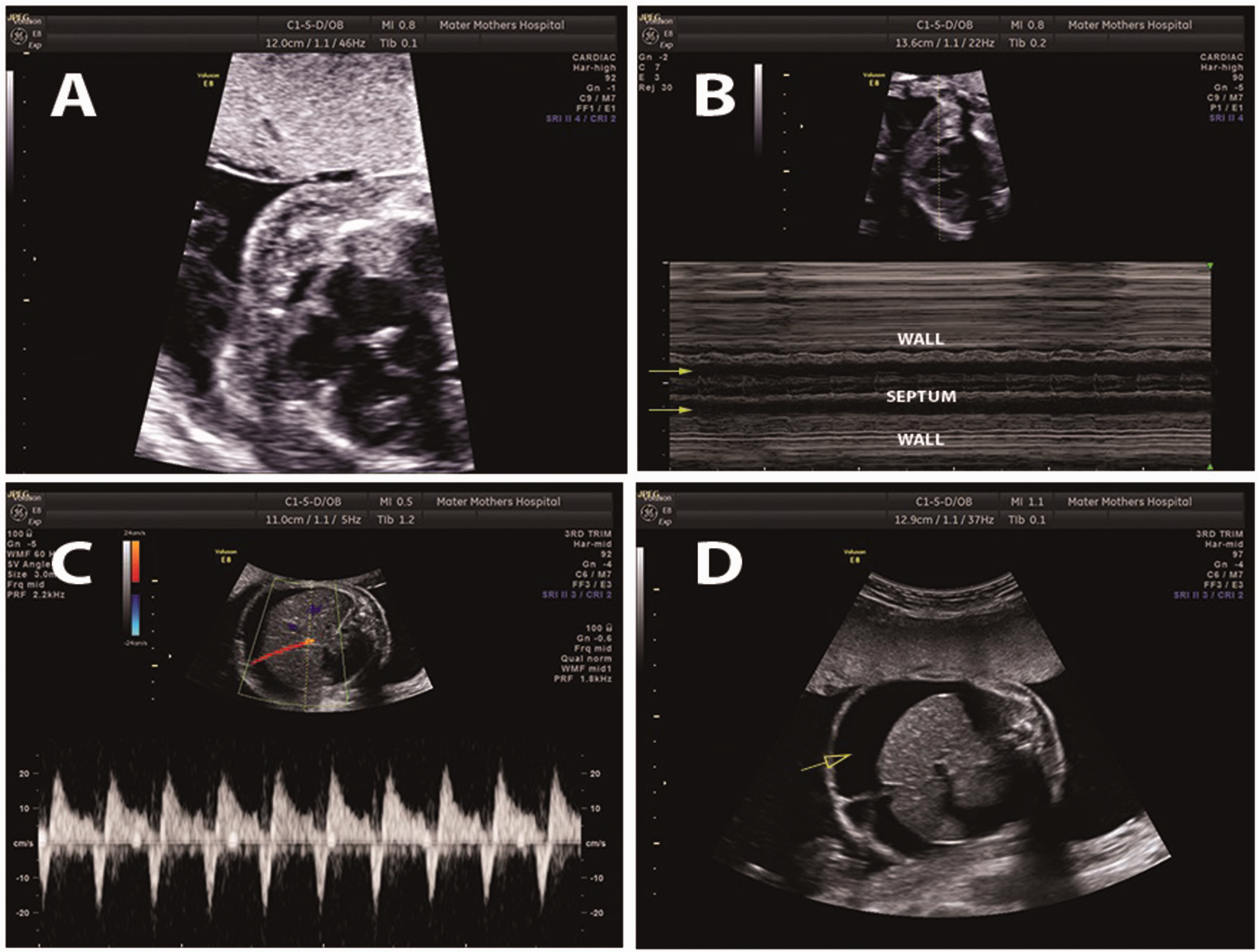
Maternal investigations taken at 24 weeks gestation indicated a normal full blood count. The blood Group was O with Rhesus Type negative and no serological evidence of anti-Rhesus antibody or recent CMV, parvovirus, rubella, or toxoplasmosis infection.
The poor prognosis with likely in utero demise of the fetus was discussed with the parents. Induction of labor was performed at 26 weeks gestation. The fetal heart beat ceased during labor and a stillborn male was delivered.
At autopsy, the male fetus was noted to be nondysmorphic, with normal growth parameters. There was severe hydrops, with skin edema, a large quantity of ascitic fluid (145 ml), a pericardial effusion (4 ml), and bilateral pleural effusions. The birth weight was >90th centile for gestational age, reflecting the hydrops. The heart was enlarged, weighing 9.3 g (5th–95th percentile: 4.4–9.0 g). The right ventricular thickness was 3 mm. The left ventricle showed prominent trabecular grooves, extending deeply into the wall and the left ventricular thickness was 7 mm. The ductus arteriosus was patent. There were no further anomalies of the cardiovascular system. The liver appeared congested and weighted 63.1 g (5th–95th percentile: 29.7–55.7 g), consistent with cardiac failure. There were no further anatomical abnormalities.
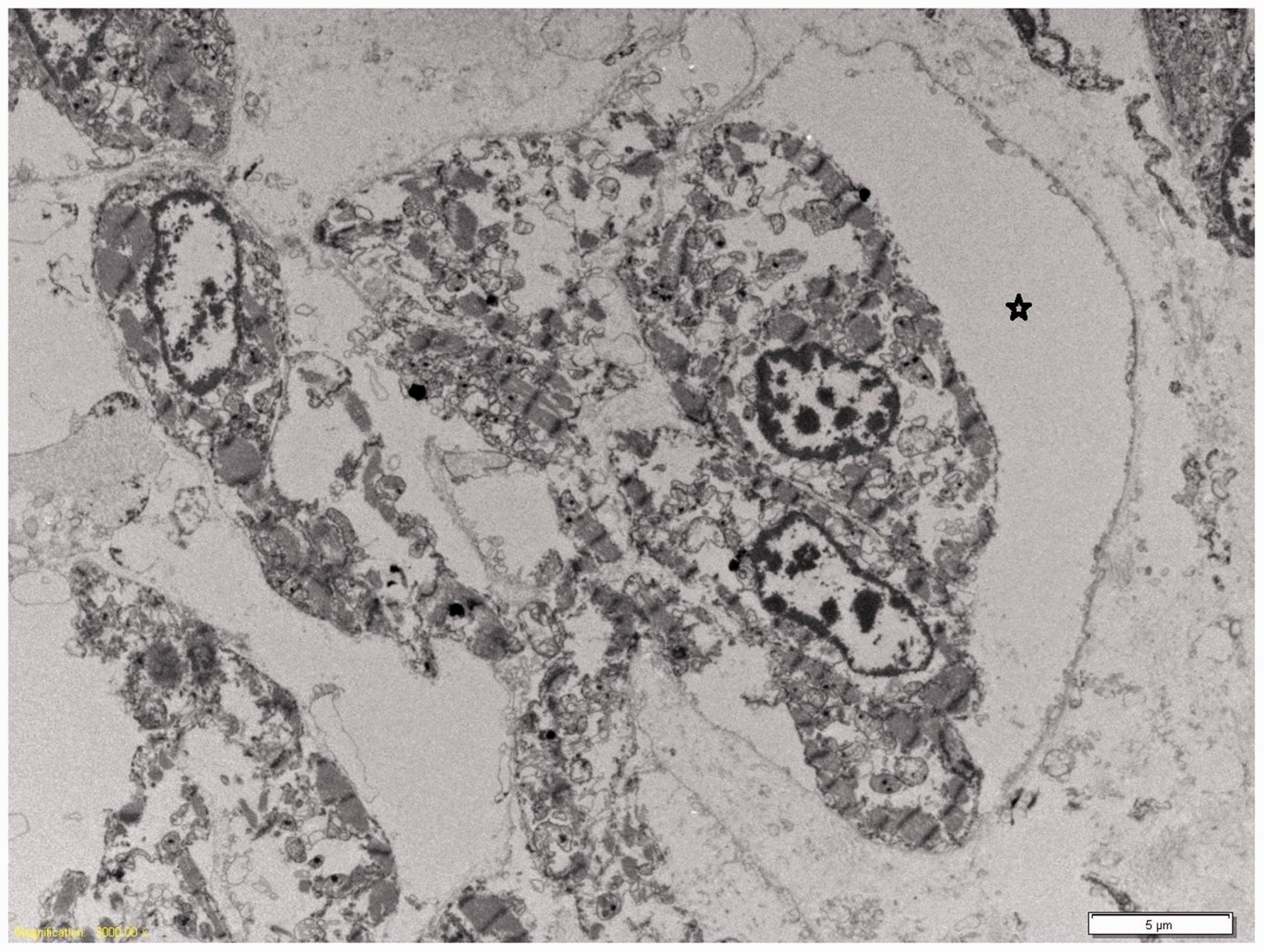
Histological sections of the left ventricle confirmed excessive trabeculation, with a thick trabecular layer compared to the compact layer (4.5 mm and 1.8 mm, respectively, giving a ratio of 2.5; Figure 2). The left ventricular endocardium, including the trabeculations, was lined by fibroelastotic endocardium (Figure 2). The myocytes surrounding the trabeculae had a swirling, disorganized arrangement, whereas the myocytes of the compact layer of the ventricular wall were more orderly and compacted. There was also endocardial fibroelastosis and trabeculation of the right ventricle, although this trabeculation was less striking than in the left ventricle. Viral inclusions were not identified, and there was no inflammation of the myocardium. Electron microscopy of the cardiac muscle showed large cardiac sinusoids, as previously documented in VNC (Figure 3).
6
However, while postmortem degeneration was noted, there was no obvious disruption of the normal parallel orientation between the sarcomeres and mitochondria. Viral cultures were performed on the pericardial fluid and were negative. Isolated left ventricular noncompaction leading to cardiac failure and severe nonimmune hydrops was diagnosed.
Photomicrograph of left ventricle showing the comparative thickness of the inner trabecular layer (right, 4.5 mm) to the outer compact layer (left, 1.8 mm), together with the complex, excessive trabeculae lined by fibroelastotic endocardium (Masson’s Trichrome; original magnification ×2). Transmission electron microscopy of heart showing large cardiac sinusoids (*). Original magnification ×4000.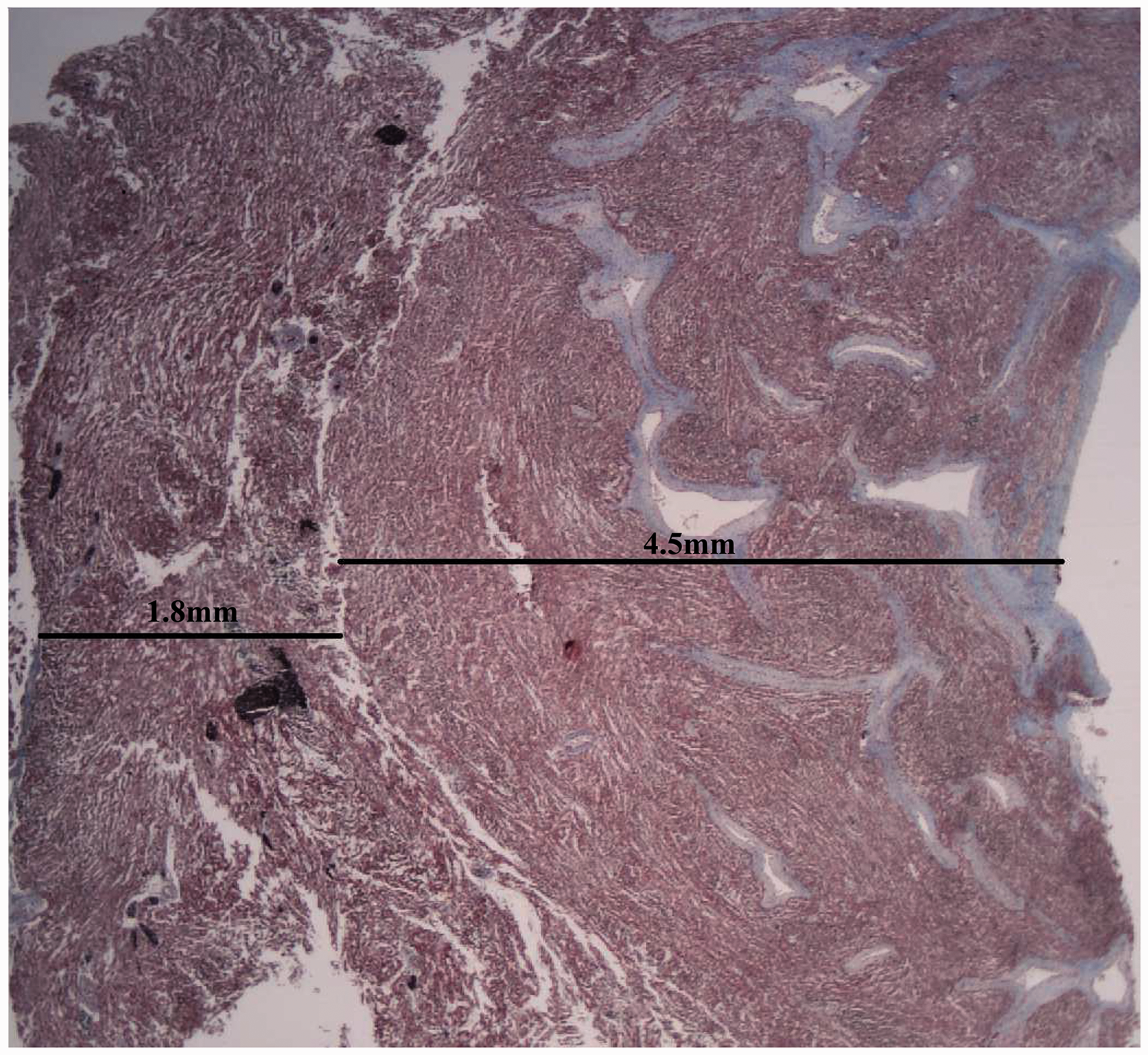
Affymetrix CytoScan750K SNP array analysis of DNA from postmortem liver (fresh frozen) revealed a normal male karyotype: arr(1-22)x2,(XY)x1, together with two long contiguous stretches of homozygosity (LCSH), one of which encompassed the MYBPC3 gene.
A 65-gene Illumina Next Generation Sequencing Cardiomyopathy panel screen revealed three variants of note in the fetus. The mode of inheritance of these three variants could be assessed, since the genetic sequence of these three genes was available in the parents via a separate research initiative (data not shown), in which whole genome sequencing had been performed on parental DNA (BGI-Complete Genomics Blackbird, Mountain View, CA, USA). The three variants of note which were identified in the fetus are as follows:
A heterozygous missense substitution Variant of Uncertain Significance (VOUS) in MYH6: NM_002471.3 (MYH6): c.2579G > A in exon 21, NP_002462.2 (MYH6): p. (Arg860His). This was maternally inherited. The affected amino acid is highly conserved and was designated a VOUS (Alamut Visual database v.2.7.1 on 30/12/2015) on the basis of conflicting in silico software prediction analyses. It has been observed previously in the context of a patient who suffered a cardiac arrest and is present in SNP databases at a frequency of <1%. A heterozygous missense substitution VOUS in TNNC1: NM_003280.2 (TNNC1): c.262G>A in exon 4, NP_003271.1 (TNNC1): p. (Asp88Asn). This was a de novo mutation (ie, not found in either maternal or paternal DNA). The affected amino acid is moderately conserved and is situated in the flexible linker domain region that connects the regulatory and structural parts of the protein. It was designated a VOUS (Alamut Visual database v.2.7.1 on 30/12/2015) on the basis of conflicting in silico software prediction analyses. It has not been reported in other clinical cases, as well as not being present in SNP databases. A combination of alterations in MYBPC3:
Homozygosity over the entire gene (detected by Affymetrix 750 K SNP array analysis). The region of homozygosity spanned 46,497,427 – 51,550,787 (5 Mb). A homozygous missense substitution: c.706A > G, p. (Ser236Gly). This variant was found in both the maternal and paternal DNA, and since it was seen within an area of genomic homozygosity within the fetus, the mode of inheritance could not be evaluated. This is a weakly conserved amino acid that is present in SNP databases, with widely varying allele frequencies when comparing the North European and Japanese populations. In the heterozygous form, it would be considered likely benign, due to its population prevalence. In the homozygous form, it might be classified as a VOUS, due to the rare occurrence of individuals homozygous for this variant, and conflicting in silico software prediction analyses.
Discussion
VNC can present as progressive cardiac failure in fetal through to adult life. Presentation in the fetus is rare and can be documented from the second trimester onwards, with no gender bias, variable presentation, and outcome.7,8 The etiology of VNC appears multifactorial, and recently it has been suggested that the etiology of adult-onset VNC is different from that of childhood-onset disease. Adult-onset VNC has been associated with autosomal dominant inheritance. However, a significant proportion of adult-onset VNC may occur due to acquired cardiac disease. 9 VNC presenting in childhood appears predominantly genetic in etiology and has been documented to have variable modes of inheritance, including X-linked and autosomal dominant or via the mitochondrial genome. 9 To date, mutations in genes encoding proteins involved in a wide variety of cellular functions have been associated with VNC: the Taffazin gene (TAZ/G4.5) which encodes a protein component of the mitochondrial electron transport chain; α-dystrobrevin (DTNA), which forms part of the dystrophin-associated glycoprotein complex important in membrane stability during muscle activity; and those encoding cytoskeletal proteins (LMNA and ZASP/LDB3) and lamin A/C. 9 There is also evidence that mutations in genes encoding sarcomeric proteins previously implicated in hypertrophic cardiomyopathy (HCM; eg, MYH7, TNNT2, and ACTC) are associated with VNC.10,11 Blasco et al. 12 reported a case of fetal HCM which reduced in postnatal life, following which VNC developed, indicating that there is morphologic overlap between these two genetically determined cardiomyopathies. Few studies of prenatally diagnosed VNC have been investigated molecularly. Notable exceptions are two recent reports, both of which have identified mutations in the sarcomeric gene MYH7.7,13
Molecular analysis of the fetal DNA in this case did not reveal previously-reported VNC-associated mutations. However, a 65-gene Illumina NGS Cardiomyopathy panel screen revealed variants of uncertain significance in MYH6, TNNC1, and MYBPC3 that encode three sarcomeric proteins: the myosin heavy chain alpha isoform, Troponin C, and myosin-binding protein-C cardiac-type proteins, respectively.
The myosin heavy chain alpha protein (MYH6) forms most of the cardiac muscle thick filament. It is highly expressed in both the atria and ventricles during embryonic development. 14 The amino acid substitution occurs in a highly conserved residue within the neck domain of the protein, a region which is known to harbor the A1004S substitution that has been associated with atrial septal defect, as well as cardiomyopathy. 15 It is interesting to note that pathogenic mutations in the related sarcomeric gene, MYH7, have been found in previously documented cases of prenatally diagnosed VNC.7,13
The Troponin C protein (TNNC1) binds to cardiac troponin I and troponin T in cardiac myocytes and plays the key role in initiating muscle contraction, when Ca2+ binds to its regulatory domain. It is also expressed during cardiac development. The substitution identified in this case occurs in the region linking the regulatory and structural domains and the biological impact of this, if any, is not known, nor has this particular gene variant been reported previously. This variant was found to have occurred de novo in the fetus and hence its impact in the heterozygous state could not be assessed by evaluating the parental phenotype.
The myosin-binding protein-C (MYBPC3), cardiac-type protein is also expressed in cardiac muscle during human development. It is required for the normal organization and function of the cardiac contractile apparatus and it binds to myosin, actin, and titin. The findings in MYBPC3 identified in this case (homozygosity for a missense substitution) is classified as VOUS and it is thus unknown what impact this effectively homozygous variant might have on sarcomeric function.
The VOUS status ascribed to the alterations found in these three genes indicates that it is not possible to ascribe a pathogenic or nonpathogenic association with the development of the VNC encountered in this case. Further, it is not known whether the combination of variants in any two of these three genes, or in all three genes, could act synergistically to impact negatively on cardiac development and function. However, that possibility cannot be excluded, particularly given the key role played by each gene in the development and functioning of the embryonic heart.
Variable ultrastructural anomalies have been documented in VNC. Siew and Markey (2011) documented persistent dilated embryonal sinusoids by electron microscopy in two cases of LVNC (a girl of 10 years and a 5-month-old boy) and a similar appearance was noted in our case. 6 Alternatively, Liu et al. 16 documented disorganized sarcomeric and mitochondrial organization in the hearts of patients with VNC (age range 13–35 years), together with abnormal mitochondrial structure. 16 While our electron microscopic findings were compromised due to postmortem autolysis, similar disorganization of sarcomeric and mitochondrial alignment was not seen in the current case.
Conclusion
We present a case of isolated left VNC detected as fetal hydrops at 24 weeks gestation. Review of the literature indicates that VNC presenting in the fetus is rare, and that the outcome is variable. However, VNC leading to fetal hydrops appears to be associated with a high rate of fetal or neonatal demise. We utilized a genomic approach to the investigation of this case of VNC, identifying VOUS in three sarcomeric proteins. Given that the contractile apparatus relies on the interaction of multiple proteins, coupled with the knowledge that mutations in all of these three proteins cause various cardiac pathologies, one can postulate that in this case of VNC, cardiac function was critically compromised due to a synergistic impact of these variants on sarcomeric function and development. However, despite the above hypothesis, a conventionally definitive genetic cause of the VNC in this case remains unresolved.
Footnotes
Acknowledgments
We thank Prof. Dr. Med. Wolfgang Rottbauer, Universitätsklinikum Ulm, Ulm, Germany and Prof. Shirley Siew, Michigan State University, East Lansing, Michigan, USA for providing helpful advice on our findings and Naomi McCallum, Queensland Health, Australia for her electron microscopy expertise.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.