
Abstract
Alpha thalassemia major is a hemoglobinopathy caused by the inactivation or deletion of all 4 α-globin alleles. We describe a case of α-thalassemia major with atypical ultrasound and neuropathological findings. The mother had her first prenatal visit at 27 4/7 gestational weeks. Ultrasound revealed a hydropic fetus with multiple anomalies. However, the middle cerebral artery peak systolic velocity (MCA-PSV) suggested that the likelihood of fetal anemia was low. Given the poor prognosis of hydrops fetalis, the parents opted for termination of pregnancy. The neonate died shortly after birth. Autopsy revealed a markedly hydropic female infant with severe limb reduction defects and, in contrast to what was suggested by the prenatal MCA-PSV measurement, unequivocal signs of severe anemia. The brain showed diffuse white matter gliosis. Genetic testing subsequently identified HBA1 and HBA2 deletions, consistent with α-thalassemia major. This case highlights the potential pitfall of MCA-PSV, which is nowadays considered the gold standard for noninvasive detection of fetal anemia. In addition, this is 1 of 2 published case reports detailing neuropathological findings in a fetus or neonate with α-thalassemia major and the first to link α-thalassemia major with diffuse white matter gliosis.
Keywords
Introduction
Alpha thalassemia (α-thalassemia) is one of the most common human monogenic diseases. 1 It is an inherited hemoglobin disorder that results from defects in the HBA1 and/or HBA2 genes (16p13.3), both of which code for the α-globin chain, a subunit of hemoglobin. Patients with α-thalassemia have a reduced production of α-globin chains. 2 Since each of the HBA1 and HBA2 genes has 2 alleles, there are a total of 4 alleles that produce the α-globin chains. The different types of α-thalassemia arise from defects in some or all of these alleles. The most severe variant, α-thalassemia major, occurs when all 4 alleles are nonfunctional.
Fetal hemoglobin (HbF) is the predominant hemoglobin throughout most of fetal life. Typically, a fetus begins production of HbF at approximately 6 weeks gestational age (GA), replacing the embryonic hemoglobins (Hbs Gower I, Gower II, and Portland). HbF is composed of 2 α- and 2 γ-globin chains. When both HBA1 and HBA2 are nonfunctional, as in α-thalassemia major, the excess γ-globin chains bind to form a tetramer, called Hb Barts, which has an unusually high affinity for oxygen and is unable to efficiently deliver oxygen to fetal tissues. Virtually all fetuses with this condition demonstrate fetal anemia and hydrops. 3
Herein, we present a case of α-thalassemia major with atypical prenatal ultrasound presentation and novel neuropathological findings.
Case Report
A 27-year-old gravida 1, para 0 woman presented at our hospital at 27 4/7 weeks GA with worsening right upper quadrant pain for the past 2 weeks. Her medical and family history was unremarkable. She and her partner were nonconsanguineous.
The couple had recently migrated from China, where she had her initial prenatal care. The partner had α-thalassemia trait (2 α-globin gene deletions in cis), but the patient had never been worked up for α-thalassemia. Her hemoglobin and MCV were low, at 91 g/L (normal range: 120–150 g/L) and 69 fL (normal range: 80–96 fL), respectively.
At the time of presentation, the estimated fetal weight and abdominal circumference were well above the 99th percentile. The head and femur measurements were at the third and first percentile, respectively. Cardiothoracic ratio was over 99th percentile. Skin edema and hepatomegaly were noted. No fetal movement or tone was detected. The placenta was enlarged and hydropic.
The middle cerebral artery (MCA) peak systolic velocity (PSV) was 1.35 multiples of median (MoM), suggesting a low risk of moderate to severe anemia. The umbilical artery and MCA pulsatility index Doppler measurements were normal with no features of cerebral redistribution.
Given the poor prognosis of fetal hydrops, the parents opted for termination of pregnancy and induction of labor. The infant lived for 1 h and 27 min after birth. A full autopsy was requested.
Autopsy revealed a premature phenotypically female infant, with diffuse skin edema and a markedly distended abdomen (Figure 1). Crown-heel and crown-rump lengths were 39.5 cm (expected mean: 35.1 ± 3.0 cm) and 28 cm (expected mean: 25.0 ± 2.1 cm), respectively. The infant had a puffy face, a small nose, ears with simple outer helices, and short limbs with striking anomalies (Figure 1). All digits on the left hand were short. On the right hand, the second to fifth digits were hypoplastic and rudimentary, and a wide gap was present between the first and second digits. All 10 toes were hypoplastic, and wide gaps were present between the first and second digits on both feet. Toenails were not well formed.
A 27 4/7-gestational week neonate with alpha thalassemia major. A, The neonate was hydropic and had a puffy face and relatively short limbs. B and C, The hand and foot showed markedly abnormal development. Note: Color image is available in the online version of the article.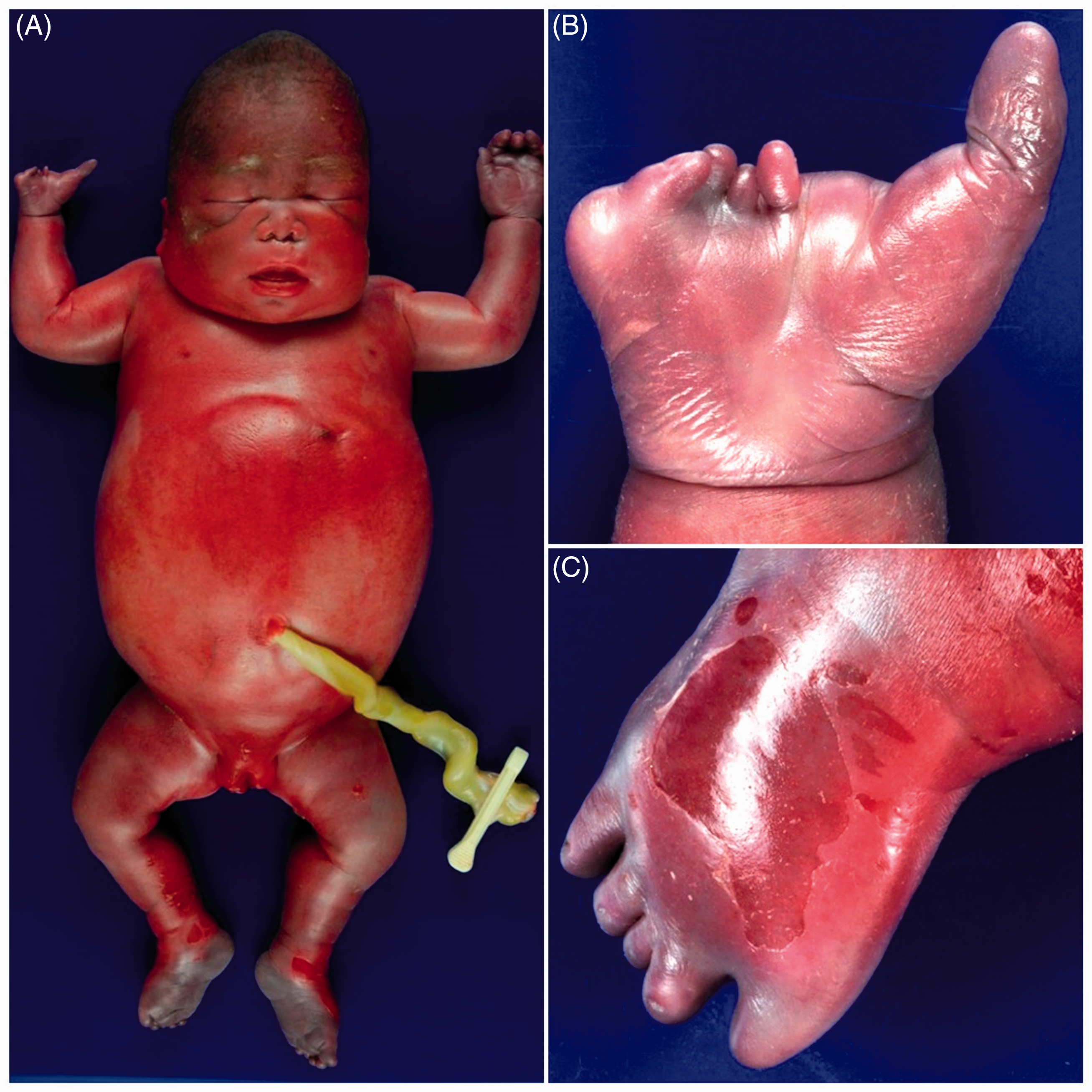
Internal examination revealed pleural, pericardial, and peritoneal effusions, a small thymus (1.44 g, expected mean: 2.85 ± 1.22 g), cardiomegaly (16.2 g, expected mean: 7.13 ± 2.11 g), bilateral pulmonary hypoplasia (8 g, expected mean: 22.7 ± 7.3 g), hepatomegaly (157 g, expected mean: 42.6 ± 11.5 g), splenomegaly (3.86 g, expected mean: 2.26 ± 0.96 g), short small (65 cm, expected mean: 150 cm), and large (17 cm, expected mean: 30 cm) intestines. Rotation of the bowel was incomplete. A large atrial septal defect and coarctation of the aorta distal to the left subclavian artery were noted. Microscopic examination showed increased extramedullary hematopoiesis in the liver, kidneys, and pancreas.
The brain weighed 144 g (expected mean: 135 ± 24 g). The cerebrum and cerebellum were congested, and prominent vessels were present in the perisylvian region bilaterally (Figure 2(A)). Gyration pattern was normal. No malformations, cavitations, or kernicterus were seen. Microscopically, the cortical lamination was normal and appropriate for GA. The cerebral vessels were severely congested and contained numerous nucleated red blood cells. Severe vascular congestion and small hemorrhages were present in the germinal matrix. The cerebral white matter was diffusely gliotic with extensive reactive astrocytes, perivascular calcifications, multinucleated giant cells, and rare gliotic cavities (Figure 2(B) to 2(I)). Scattered perivascular calcifications with multinucleated giant cell reaction and reactive astrogliosis were also noted in the globus pallidus. These findings were consistent with diffuse white matter gliosis. No evidence of pontosubicular necrosis or infection was found.
Macroscopic and microscopic images of brain in a 27 4/7-gestational week neonate with alpha thalassemia major. A, The brain was congested with prominent tortuous vessels on the external surface. B, The white matter was diffusely gliotic. C, Extensive reactive astrocytosis is present. D and E, GFAP immunolabeling highlights the reactive astrocytes. F, Many nucleated red blood cells were noted within the capillaries. F–H, Scattered foci of perivascular calcifications associated with multinucleated giant cells were present. I, CD68 immunostain is weakly positive in the multinucleated giant cells. Note: Color image is available in the online version of the article.
The placenta was extremely large (923 g, expected mean: 270 g) and pale, with diffuse villous edema, massively increased fetal nucleated red blood cells, and erythroblasts in the fetal vessels.
The mother subsequently underwent genetic testing and, like her partner, was found to have α-thalassemia trait (2 α-globin gene deletions in cis). Array CGH/chromosomal microarray analysis performed on the infant’s DNA using the CytoSureTM ISCA 8X60K v2.0 platform (Oxford Gene Technology) identified a 2 copy number loss spanning 30 probes representing a 16 kb region within the short arm of chromosome 16 from nucleotide 216,630 to 232,684. This deleted region encompasses the HBM, HBA1, HBA2, and HBq1 loci associated with α-thalassemia major.
The couple was counseled, and eventually had a successful second pregnancy, delivering a term healthy neonate with α-thalassemia carrier status.
Discussion
The absence of α-globin chains led to ineffective erythropoiesis and profound anemia, prompting marked extramedullary hematopoiesis in the liver. The severe anemia also generated a high output cardiac state, leading to cardiomegaly, heart failure, fetal hydrops, and placentomegaly. Although cardiomyopathy, cardiomegaly, and atrial septal defect have been described in cases of α-thalassemia major, aortic coarctation has not been previously reported.
Hb Barts, having a high affinity for oxygen, prevents release of oxygen to fetal tissues, causing severe tissue hypoxia. The limb reduction defects seen here have been previously described in fetuses with α-thalassemia major4,5 and are believed to be vascular disruptive phenomena secondary to early-onset chronic hypoxic state. Indeed, a high incidence of limb defects were noted in offspring of mice subjected to regular blood sampling that induced hypovolemia and hypoxemia. 6 In addition, limb defects have been detected on ultrasound in fetuses with α-thalassemia major as early as 10 weeks GA, when the fetus’s primary form of hemoglobin switches from embryonic hemoglobins to HbF. 7
Diffuse white matter gliosis is thought to represent hypoxic-ischemic damage to vulnerable cerebral white matter on a spectrum with periventricular leukomalacia. In this case, the damage is likely secondary to chronic hypoxia in the setting of α-thalassemia major, although this association has not been reported in the literature previously.
Although rare, α-thalassemia major is a well-documented disease; therefore, it is rather unexpected when we could only identify 2 published papers with morphologic descriptions of brain associated with α-thalassemia major, and only one of them contained microscopic descriptions of the brain. Liang et al. reported microcephaly and hydrocephalus and Adam et al. found periventricular heterotopias in a fetus with α-thalassemia major.4,8
In the past, α-thalassemia major was invariably a fatal condition, but medical advances over the past few decades have permitted the survival of a small number of affected patients. Chan et al. identified 9 survivors and found that 2 had long-term neurological deficits. 9 Songdej et al. studied an international registry of survivors with α-thalassemia major. Neurodevelopmental outcomes information was available in 55 patients. 10 Although most patients in the cohort were either developmentally normal or had only mild developmental delay, 11 patients suffered significant adverse neurodevelopmental outcomes. These 2 studies highlighted that neurologic abnormalities are not uncommon in survivors with α-thalassemia major. However, the authors did not speculate the reasons behind the neurologic deficits, and radiologic or pathologic brain findings were not available in either paper. With our index case of diffuse white matter gliosis in a neonate with α-thalassemia major, we speculate that some of these α-thalassemia major survivors might have sustained similar chronic ischemic brain injury during fetal development that persisted into the postnatal period and adulthood.
The prenatal ultrasound presentation was also curious. MCA-PSV is considered to be the gold standard for noninvasive detection of moderate to severe anemia. In this case, the MCA-PSV was only 1.35 MoM, which suggested a low risk of moderate to severe fetal anemia. Yet, autopsy demonstrated unequivocal evidence of severe anemia. This discrepancy has been previously noted in a few cases of α-thalassemia major.11,12 Leung et al. speculated that the discrepancy could be due to the relatively high hematocrit levels of fetuses with α-thalassemia major and to the differences in physical properties between Hb Barts and HbF. 11 In cases of suspected α-thalassemia major, it has been suggested that cardiothoracic ratio in combination with MCA-PSV could increase the sensitivity for fetal anemia detection.11,12
Conclusion
In summary, this case highlights the potential pitfall of using MCA-PSV measurement in diagnosing anemia in fetuses affected by α-thalassemia major. In suspected cases of α-thalassemia major, combining fetal cardiothoracic ratio measurement with MCA-PSV has been shown to increase detection rate of fetal anemia. Furthermore, to the best of our knowledge, this case represents the second published report of neuropathology in a fetus/neonate with α-thalassemia major and the first to make the association between α-thalassemia major and diffuse white matter gliosis, which is believed to be manifestation of chronic ischemic brain injury.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.