
Abstract
Pediatric glial tumors are unique from their adult counterparts. This important distinction is recognized and incorporated into the World Health Organization classification of central nervous system tumors and applies to both high- and low-grade gliomas, incorporating their specific molecular profiles. Molecular alterations in pediatric high-grade gliomas provide important prognostic information, for example in H3 K27M-mutant tumors. The integration of molecular information is also important for pediatric low-grade gliomas due to their overlapping morphologies and the prognostic and therapeutic implications of these molecular alterations. In this paper, we cover a variety of glial tumors, encompassing neoplasms with predominantly glial histology, astrocytic tumors, oligodendroglial tumors, and mixed glioneuronal tumors. Considering the complexity of this evolving field, the purpose of this article is to offer a practical approach to the diagnosis of pediatric gliomas, including the selection of the most appropriate molecular surrogate immunohistochemical stains, basic molecular studies, and more sophisticated techniques if needed. The goal is to reach a rapid, sound diagnosis, helping guide clinical decision-making regarding prognosis and potential therapies.
Introduction
The World Health Organization (WHO) 2016 classification of central nervous system (CNS) tumors recognized, for the first time, that many high-grade glial tumors in children are distinct from their adult counterparts. 1 Subsequent publications and updates have expanded this important distinction to low-grade gliomas as well.2–5 The 2021 WHO classification of tumors of the CNS confirms pediatric gliomas as unique entities with specific molecular profiles. 6
The integration of histology and molecular information into the diagnostic line of brain tumors (the “integrated diagnosis”) was introduced in the 2016 WHO classification and expanded in the fifth edition. 1 , 6 Molecular alterations in pediatric high-grade gliomas (pHGG) provide important prognostic information, for example in H3 K27M-mutant tumors. This integration of molecular information is also important for pediatric low-grade gliomas (pLGG) due to their overlapping morphologies and the prognostic and therapeutic implications of these molecular alterations, as discussed below.
In this paper, we will cover a variety of glial tumors, encompassing neoplasms with predominantly glial histology, astrocytic tumors, oligodendroglial tumors, and mixed glioneuronal tumors. Ependymal tumors will be discussed separately (please refer to the chapter on ependymal tumors by Santi et al.). For consistency with the 2021 WHO classification, Arabic numerals will be used for tumor grades. Tumors will be divided into three sections: pLGG (WHO Grades 1 and 2), pHGG (WHO grades 3 and 4), and infantile gliomas.
Considering the complexity of this evolving field, the aim of this paper is to offer a practical approach to the practicing pediatric pathologist, including the selection of the most appropriate molecular surrogate immunohistochemical stains, basic molecular studies, and more sophisticated techniques if needed. The goal is to reach a rapid, sound diagnosis, helping guide clinical decision-making regarding prognosis and potential therapies.
Pediatric Low-Grade Gliomas
pLGG are the most frequent brain tumors in children, accounting for approximately 30% of all cases. 7 , 8 These represent a diverse group of tumors of varied histologies and cell types. pLGG can be found throughout the neuroaxis including the cerebral hemispheres, posterior fossa and spinal cord, and some entities may involve of the leptomeninges. While many pLGG appear radiographically circumscribed, there is often some microscopic degree of infiltration of brain parenchyma near the tumor edges. Additionally, some pLGG are diffusely infiltrative in nature, such as diffuse astrocytomas (DA). Classification of pLGG often requires integration of clinical, radiologic, histologic, and molecular information. However, it should be noted that a subset pLGG do not fit into a well-defined WHO category. For such tumors, a histologic diagnosis of pLGG, not otherwise specified (NOS) or not elsewhere classified (NEC) may be appropriate. Specifically, the NOS designation is given to tumors for which genetic testing has not been performed and for tumors where such diagnostic testing failed. On the other hand, a NEC diagnosis is utilized for tumors when the necessary molecular tests “were performed and results are available, but in which those results do not allow for a specific WHO diagnosis”. 9
Morphologic Entities
pLGG are a morphologically diverse group of tumors encompassing those of astrocytic, oligodendroglial, glial, and mixed glioneuronal histologies. Examples of tumor histologies can be found in Figure 1, and brief descriptions of typical tumor histologies are presented in Table 1. There may be significant overlap in morphology amongst the various tumor types (for example, dysembryoplastic neuroepithelial tumors (DNET) and oligodendrogliomas, as discussed below). To further complicate matters, pLGG can demonstrate morphologic features of more than one tumor type. pLGG may distinguished from their high-grade counterparts on the basis of specific morphologic features or, for certain tumors such as DA, by the absence of microvascular proliferation, necrosis, and increased mitotic activity. While most pLGG do not progress to higher grades, some entities such as pleomorphic xanthoastrocytomas (PXA) may undergo anaplastic transformation. Both histologic features (such as increased mitotic activity) and molecular findings (see discussion of CDKN2A deletion below) can help to identify these occurrences.

Histology of pediatric low-grade gliomas. A, Pilocytic astrocytoma demonstrating biphasic morphology (100X magnification). B, Diffuse astrocytoma characterized by infiltrating astrocytic cells with mild nuclear atypia, lacking increased mitotic activity, microvascular proliferation, and necrosis (200x magnification). C, Pleomorphic xanthoastrocytoma which contains neoplastic cells notable for their pleomorphism as well as multinucleated giant cells. Lymphocytic inflammation is present (400X magnification). A cell with xanthomatous change is shown in the inset (600X magnification). D, Angiocentric glioma composed of monomorphic spindle cells arranged around blood vessels (200X magnification). E. Ganglioglioma containing neoplastic glial cells as well as dysplastic neurons. Numerous eosinophilic granular bodies are present (100X magnification). F, Dysembryoplastic neuroepithelial tumor composed of small, oligodendrocyte-like cells and neurons “floating” in a myxoid background (200X magnification).
Dominant Histologies and Molecular Alterations Commonly Found in Pediatric Low-Grade Gliomas.

aThis table is meant to provide an overview of the most common molecular alterations for each tumor type; this is not an exhaustive list and other alterations may be seen. For a more detailed review, please refer to Table 1 in Ryall et al. 4
bThe presence of the H3.3 p.K27M mutation in a DA warrants a diagnosis of diffuse midline glioma, WHO grade 4 provided the tumor is located in the midline (please refer to cIMPACT-NOW update 2 criteria). 43
cIDH1/2 mutations in diffuse astrocytomas and oligodendrogliomas are seen adult-type tumors. For diffuse astrocytomas, IDH-mutant, co-occurring mutations in TP53 and ATRX are often present. For adult-type oligodendrogliomas, the 1p/19q-codeletion is present in addition to the IDH1/2 mutation.
PLNTY: polymorphous low-grade neuroepithelial tumor of the young; SNV: single nucleotide variant.
The most commonly encountered pLGG is the pilocytic astrocytoma (PA), WHO grade 1, which accounts for 15.5% of all childhood primary brain tumors. 7 These astrocytic tumors may be found throughout the neuroaxis though most commonly present as cystic and solid circumscribed masses within the posterior fossa. Other low-grade astrocytomas that present in the pediatric population include pilomyxoid astrocytoma (a subtype of PA with greater tendency for local recurrence and dissemination), DA, PXA, and subependymal giant cell astrocytoma. It should be noted that DA in children are distinct from the IDH-mutant DA presenting in older children and adults and have significantly different biologic behavior. The molecular findings distinguishing these two groups of DA will be discussed in the section on Molecular Findings.
Glioneuronal tumors are composed of both glial and neuronal elements. This includes neoplasms such as ganglioglioma, DNET, desmoplastic infantile astrocytoma/ganglioglioma, papillary glioneuronal tumor, rosette-forming glioneuronal tumor, and polymorphous low-grade neuroepithelial tumor of the young (PLNTY). For tumors with WHO grades, glioneuronal tumors correspond to grade 1. Glioneuronal tumors can show histologic overlap with each other and with other types of pLGG. For example, gangliogliomas are often heterogenous and may share histologic features with PXA, PA, DA, DNET, and oligodendrogliomas. Likewise, PLNTY, a new tumor entity, are classically polymorphous, as their name suggests, and show overlapping histologic features with other pLGG. 10 Radiographic features, immunohistochemical stains and, in some cases, molecular findings may help in the diagnosis of these glioneuronal tumors.
Oligodendrogliomas present a unique diagnostic challenge in the pediatric population. Many pLGG can have oligodendroglioma-like morphology, including but not limited to, DNET, PA, gangliogliomas, and PLNTY. For tumors with oligodendroglioma morphology that lack an IDH mutation and 1p/19q-codeletion, such histologic mimics should be excluded. Similar to pediatric DA, pediatric oligodendrogliomas are biologically distinct from their IDH-mutant young adult and adult counterparts.
Angiocentric gliomas are glial tumors that correspond to WHO grade 1. While these tumors often appear well-circumscribed on imaging, they may show infiltrative growth microscopically. Angiocentric gliomas are a rare example of a pLGG which is characterized by molecular alterations which are not part of the RAS/MAPK pathway; these tumors are strongly associated with MYB alterations (see below).
Molecular Findings in pLGG
As a group, nearly all pLGG are thought to be driven by alterations that upregulate the RAS/MAPK pathway which most commonly occur as a single genetic event. 4 , 5 Even for the small subset of pLGG where a specific genetic alteration is not detected, significant upregulation of phosphorylated ERK is present. 5 Over two-thirds of pLGG have either a KIAA1549-BRAF fusion, BRAF p.V600E, and/or NF1 mutation. An illustration of the RAS/MAPK pathway alterations commonly encountered in pLGG can be found in Ryall et al. Figure 2, 4 and a summary of the molecular findings most commonly associated with various tumor types is presented in Table 1.
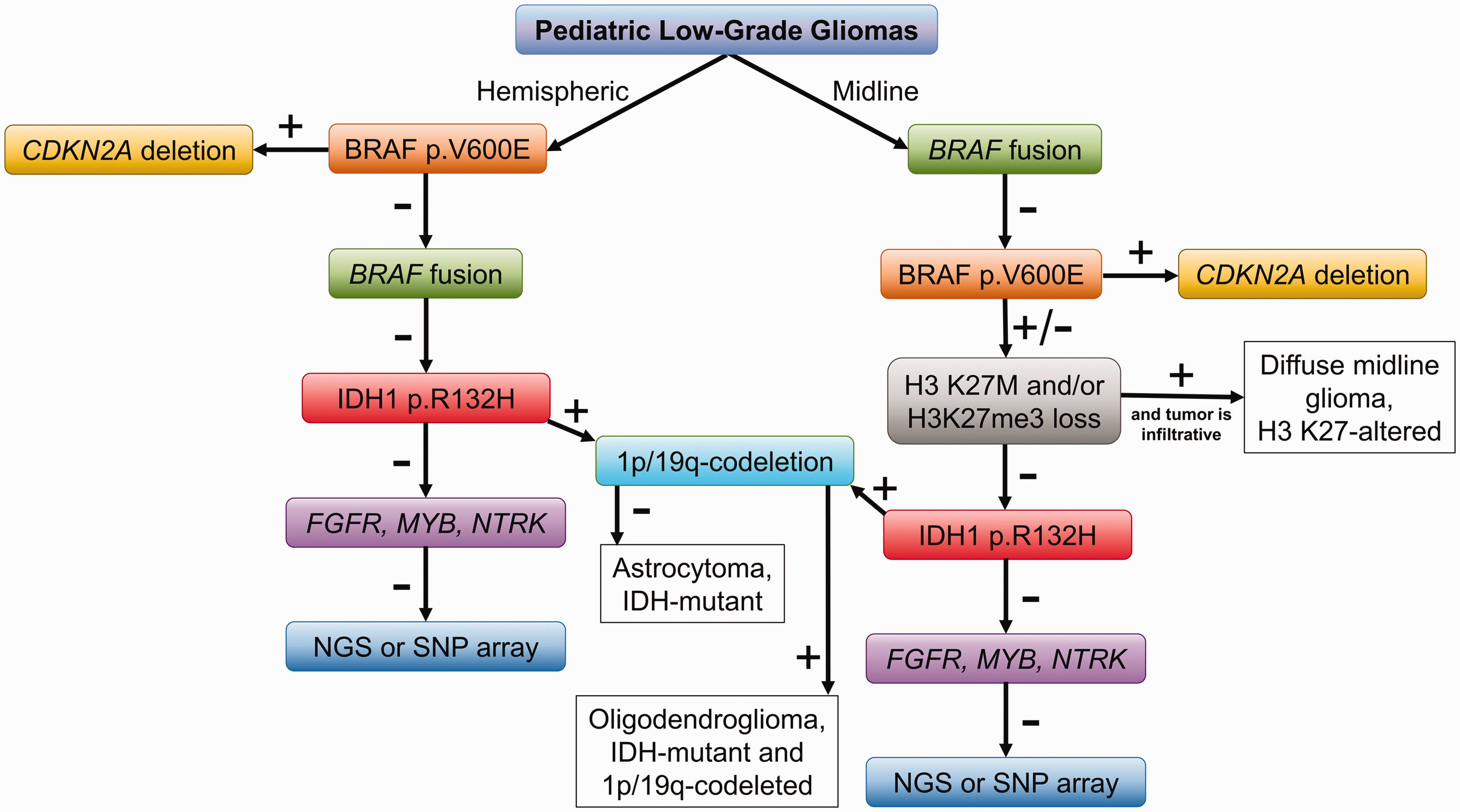
A proposed approach to the molecular characterization of pediatric low-grade gliomas. Based on tumor location (hemispheric or midline) pediatric low-grade gliomas testing for BRAF single nucleotide variants or fusions may be performed. For tumors which are positive for the BRAF p.V600E variant, testing for CDKN2A deletions is suggested. In tumors which are negative for BRAF alterations, immunostaining for H3 K27M and H3K27me3 may be done in midline tumors as well as immunostaining for IDH1 p.R132H, particularly in older children, adolescents, and young adults. If a tumor is positive for IDH1 p.R132H, follow-up testing for the 1p/19q-codeletion is suggested. In instances where these initial steps do not reveal a molecular alteration, FISH for fusions may be attempted followed by NGS or SNP array.
KIAA1549-BRAF fusions are present in up to 35% of pLGG. 11 Most frequently this fusion is present in PA of the posterior fossa but may be seen in gangliogliomas, DA, and DNET and has been reported in tumors throughout the neuroaxis (summarized in Figure 3 from Ryall et al. 4 ). This fusion results in a loss of BRAF’s regulatory domain and downstream upregulation of the RAS/MAPK pathway. KIAA1549-BRAF fusions are not found in adult-type diffuse gliomas and therefore can be helpful in confirming a diagnosis of pLGG. 12 , 13 In addition to KIAA1549, BRAF rearrangements with other fusion partners have been reported in less than 2% of pLGG; 4 ,14–16 the impact these non-canonical fusion partners have on patient outcome is currently unknown.

Histology of pediatric high-grade gliomas. A, Diffuse hemispheric glioma, H3 G34-mutant demonstrating high cellularity and palisading necrosis (100X magnification). This tumor exhibited a lack of staining for OLIG2 (B, 100X magnification), strong, diffuse p53 immunostaining (C, 200X magnification), and a loss of nuclear ATRX staining with retained staining in normal tissues including vessels (D, 200X magnification). E, High-grade, infiltrating astrocytoma of the pons showing high cellularity, nuclear atypia, and mitotic activity (200X magnification) and strong, diffuse nuclear staining for H3 K27M with negative endothelial cells serving as an internal control (F, 200X magnification), warranting a diagnosis of diffuse midline glioma, H3 K27-altered. G. Epithelioid glioblastoma characterized by sheets of tumor cells with a moderate to large amount of eosinophilic cytoplasm, distinct nuclear borders, eccentrically-located nuclei and nucleoli. This tumor contained numerous mitosis and foci of necrosis and was found to be positive for the BRAF p.V600E variant (not shown) (200X magnification). H, High-grade glioma containing scattered cells with significant nuclear pleomorphism in a patient with a germline TP53 mutation (200X magnification). The inset shows strong nuclear staining for p53 (400X magnification).
The BRAF p.V600E single nucleotide variant (SNV) can be detected in approximately 15% of pLGG. 4 This mutation renders BRAF constitutively active, resulting in upregulation of the RAS/MAPK pathway. BRAF p.V600E is found in several tumor types including PXA (40-80%), ganglioglioma (25-45%), DA (30-34%), and PA (5-10%).17–24 Tumors with the BRAF p.V600E SNV are most commonly supratentorial in contrast to those with BRAF fusions which are predominantly infratentorial (summarized in Figure 3 from Ryall et al. 4 ). pLGG with the BRAF p.V600E SNV have been shown to have worse overall survival and progression free survival compared to tumors without this alteration. 5 , 23 , 25 , 26 Additionally, for tumors with co-occurring CDKN2A deletions (most commonly seen in PXA), an increased risk of transformation to high-grade glioma has been reported. 27
NF1 mutations are seen in approximately 14% of pLGG, 5 mostly in the setting of the neurofibromatosis type 1 (NF1) tumor predisposition syndrome. NF1 mutations result in hyperactivation of RAS and the RAS/MAPK pathway. Over 80% of these NF1-associated pLGG are located in the optic pathway and have better overall survival and progression-free survival than NF1-associated gliomas arising outside the optic pathway. 5 The presence of additional molecular alterations (such as BRAF p.V600E, FGFR1, and/or H3 p.K27M) is also associated with high risk and/or recurrent tumors. Furthermore, co-occurring alterations in CDKN2A, ATRX, and TP53 may be seen in higher grade tumors. 28 Testing for the presence of additional alterations in NF1-associated pLGG can therefore provide important prognostic information and help to identify tumors that may require close observation.
Alterations in FGFR genes including mutations, fusions and tandem duplications occur in 5-10% of pLGG. 4 These alterations lead to an upregulation of the RAS/MAPK pathway and can also cause an upregulation of the PI3K/AKT/mTOR pathway (see Figure 2 from Ryall et al. 4 ). A variety of tumor types have been reported to harbor FGFR alterations, most commonly in FGFR1, including PA, DNET, gangliogliomas, pediatric oligodendrogliomas, and DA. FGFR2/3 alterations are associated with the new tumor entity, PLNTY. Co-occurring alterations are commonly seen in tumors with FGFR1 SNV but are less frequent those with tandem duplications or fusions. 15 , 16 , 29 , 30
Several other alterations affecting the RAS/MAPK pathway in pLGG have been described. These genes include NTRK, CRAF, KRAS, ALK, ROS1, MAP2K1, and PTPN11, each being seen in <2% of pLGG. For a detailed review of these alterations and additional information on those discussed above, please refer to a review by Ryall et al. 4
While these RAS/MAPK pathway-related alterations account for the vast majority of the molecular findings detected in pLGG, up to 10% of these tumors have non-MAPK or unknown alterations. 4 Non-MAPK alterations include those in the transcription factors MYB or MYBL1. MYB alterations (most commonly the MYB-QKI fusion) are seen up to 87% of angiocentric gliomas and 41% of DA with MYBL1 fusions seen most commonly in DA. 29 , 31
IDH mutations do not activate the RAS/MAPK pathway but may be seen in a small subset of pLGG. In contrast to adult lower-grade astrocytomas where IDH1/2 mutations are present in up to 70% of tumors,32–34 IDH1/2 mutations were detected in less than 1% of pLGG in a study of over 1000 tumors. 5 These IDH-mutant tumors are seen in adolescents and young adults; in one study the median age was 16 years. 35 In younger children, IDH1/2 mutations are very rare. Differentiating infiltrating gliomas as IDH-mutant and IDH-wildtype was introduced in the 2016 WHO classification. 1 In the adult population, IDH-mutant astrocytomas have significantly improved outcomes in comparison to their wildtype counterparts. This has led to some confusion regarding pediatric DA and oligodendrogliomas; in contrast to the IDH-wildtype adult gliomas, the absence of an IDH1/2 mutation in pLGG does not portend a poor prognosis. In fact, IDH-wildtype pLGG which harbor other alterations such as BRAF fusions, NF1 mutations, FGFR fusions, and MYB alterations were shown to have better overall survival and progression-free survival than IDH-mutant tumors. In one study, 50% of pLGG with IDH1 p.R132H progressed within 5 years. 5 Similar to adult tumors, pediatric DA with IDH1/2 mutations also tend to have co-occurring mutations in TP53 and ATRX, and IDH-mutant oligodendrogliomas harbor the 1p/19q-codeletion. 1 , 36 , 37 The finding of an IDH-mutant DA in the pediatric population raises the possibility of underlying mismatch repair deficiency and consideration should be given to performing immunohistochemistry to look for loss of expression of one of these proteins (MLH1, MSH2, MSH6, PMS2).
Mutations in histone H3 variants are most commonly associated with high-grade tumors and will be discussed in the pHGG section; however, their significance in the setting of pLGG bears mention. Approximately 10% of diffuse midline gliomas harboring the H3 K27M mutation are histologically DA. 1 While H3 K27M-mutant DA may have a somewhat longer overall survival than their histologically high-grade counterparts, these tumors invariably progress and have an average survival of only 1–3 years. 5 As such, these histologically low-grade DA are classified as WHO grade 4. H3 K27M mutations have also been reported in other types of pLGG including PA and ganglioglioma.38–42 These tumors do not meet criteria to be called diffuse midline gliomas (infiltrative, midline glioma with predominantly astrocytic differentiation), and therefore do not warrant an automatic WHO grade 4 designation. 43 Indeed, reports of H3 K27M-mutant pLGG with longer survivals have been reported. 5 , 38 , 39 , 41 , 42 However in a large study with long-term follow-up, all patients eventually succumbed to their disease 5 and even well-circumscribed, histologically grade 1 tumors with H3 K27M mutations have significantly worse outcomes than their wildtype counterparts. 44 pLGG with H3 K27M mutations are therefore considered high risk (see discussion below on risk stratification).
In a large study of pLGG with long-term clinical follow-up, Ryall et al. found that rearrangement-driven tumors (i.e. fusions and duplications) were associated with younger age at presentation, WHO grade 1 histology and better outcomes compared to SNV-driven tumors. 5 Tumors were further risk-stratified into three categories based on their molecular alterations. Low risk tumors included NF1-associated pLGG and those with BRAF fusions, FGFR fusions and tandem duplications, and MYB or MYBL1 alterations. These low risk tumors were found to have 20 years overall survival and progression-free survival of 96% and 58%, respectively. Intermediate risk tumors included those with SNV in BRAF (p.V600E without co-occurring CDKN2A deletion), FGFR1 and IDH1. The intermediate risk tumors progressed more frequently than low risk tumors and had 20 years overall survival and progression-free survival of 81% and 27%, respectively. High risk tumors were those with H3 K27M mutations and those with BRAF p.V600E and co-occurring CDKN2A deletions. These tumors all progressed within 10 years of follow-up (0% progression-free survival) and an overall survival at 10 years was 41%. Tumors with H3 K27M mutations had worse outcomes than those with BRAF p.V600E with co-occurring CDKN2A deletion. 5 Additional factors such as tumor location also play an important role in patient outcomes. The most favorable predictor of outcome remains complete surgical resection; infiltrative tumors and those involving midline structures are not as amenable to resection as well-circumscribed neoplasms of the cerebral hemispheres and cerebellum. 45
Not only do the molecular alterations in pLGG provide important prognostic information, they may also present opportunities for targeted therapies such as BRAF and MEK inhibitors. Several clinical trials are ongoing to investigate the use of targeted therapies in pediatric gliomas. For a detailed review of targeted therapies in pLGG, please see Ryall et al. 4
An Approach to Molecular Testing
Given the importance of molecular information in the classification, risk stratification and treatment of pLGG, various approaches to molecular testing in these tumors have been proposed. Next Generation Sequencing (NGS) panels optimized to detect common alterations in pLGG can be used as a single-method approach. However, NGS can be expensive and is not always available, in which case step-wise approaches can be employed using immunohistochemistry (IHC) stains, fluorescent in situ hybridization (FISH), polymerase chain reaction (PCR) and single nucleotide polymorphism (SNP) array. Such methods work well for pLGG as these tumors generally harbor a single molecular alteration. One approach is presented in Figure 2. As a first step for tumors located within the cerebral hemispheres, immunohistochemistry for BRAF p.V600E may be performed; for tumors which are positive, testing for CDKN2A deletion (either FISH or SNP array) identifies those tumors with deletions which are more likely to progress. For hemispheric gliomas which are negative for BRAF p.V600E by IHC, testing for BRAF fusions can be performed (FISH, PCR). For posterior fossa gliomas (especially for those that are histologically PA), testing for BRAF fusions may be performed upfront as most of these tumors harbor the KIAA1549-BRAF fusion. In the instances where posterior fossa PA are negative for BRAF fusions, BRAF p.V600E IHC is then recommended, followed-up by testing for CDKN2A deletions in tumors which are positive. With the exception of BRAF fusion-positive PA of the posterior fossa, immunostaining for H3 K27M should be performed in midline gliomas to identify high-risk tumors; if the tumor is histologically a DA, a diagnosis of diffuse midline glioma, WHO grade 4 would then be rendered. For tumors which are histologically DA or oligodendrogliomas, IDH1 p.R132H IHC should be performed, especially in tumors of older children and adolescents. For IDH-mutant tumors, 1p/19q-codeletion testing (FISH or SNP) should be pursued to identify adult-type oligodendrogliomas. This approach can be used to identify the molecular alterations in more than half of pLGG. For tumors lacking the above BRAF, IDH1, and H3 K27M alterations, additional testing can be employed. FISH may be used to identify fusions involving the FGFR1, MYB, and NTRK genes. SNP array or NGS may also be useful in such instances to identify less common alterations not detected by IHC and FISH. Additional schemes for the molecular testing in pLGG have been proposed (see Figure 4 from Ryall et al. 4 ).
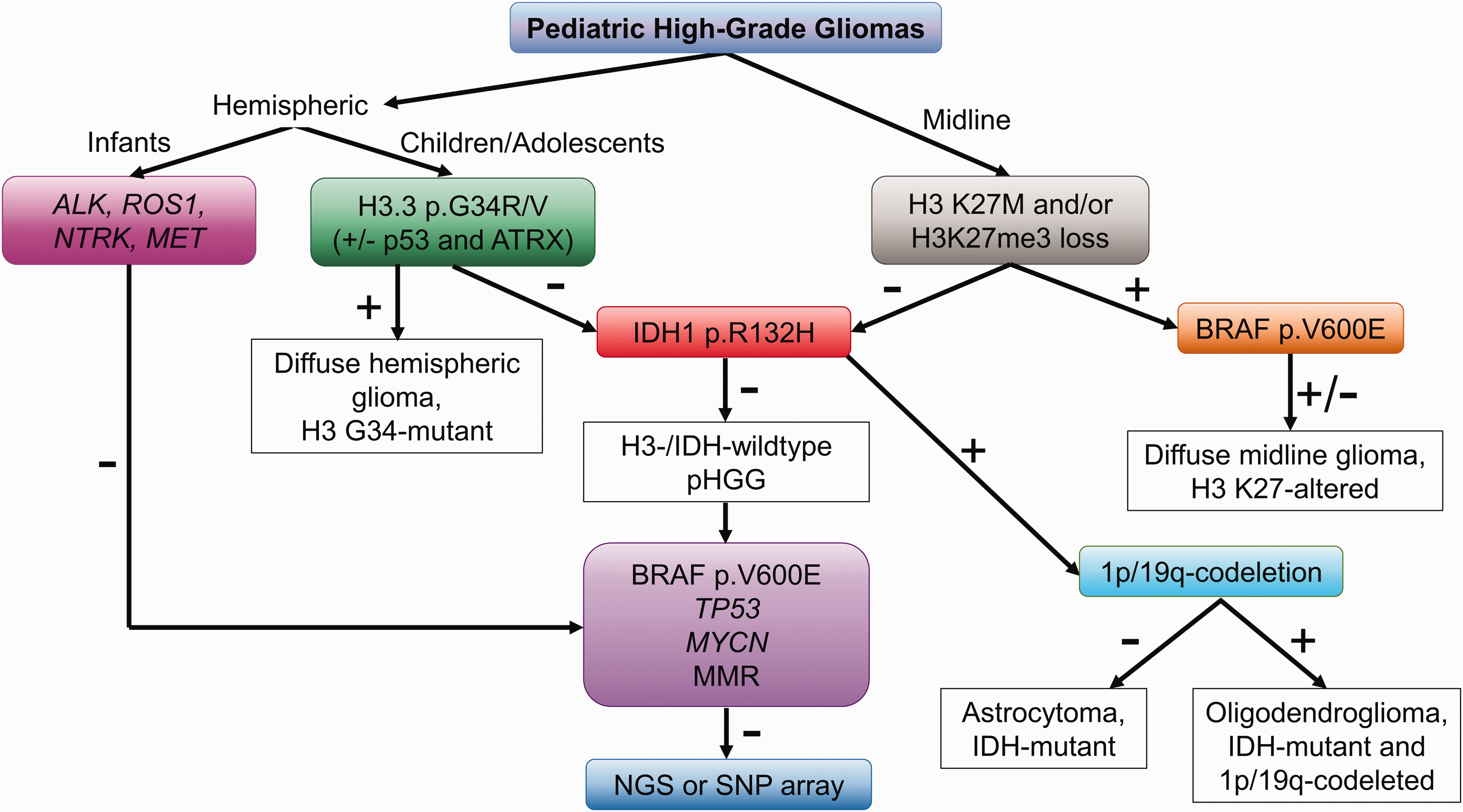
A proposed approach to the molecular characterization of pediatric high-grade gliomas. Based on tumor location, hemispheric or midline, testing for H3.3 p.G34R/V and H3 p.K27M mutations, respectively, should be performed. For midline tumors, immunostaining for H3K27me3 can also be performed to identify tumors with a loss of H3K27me3, which warrant a diagnosis of diffuse midline glioma, H3 K27-altered. In infants, testing for fusions commonly encountered in infantile high-grade gliomas should be performed in lieu of testing for H3.3 p.G34R/V. For tumors which are negative for histone alterations, immunostaining for IDH1 p.R132H should be performed, particularly in older children, adolescents, and young adults. For IDH-mutant tumors, follow-up 1p/19q-codeletion testing distinguishes astrocytomas from oligodendrogliomas. In tumors which are H3-/IDH-wildtype, testing for other common driver alterations may be considered followed by NGS or SNP array.
Upon completion of molecular testing, a unified diagnosis for pLGG can be given in a layered format. For example, the report for a midline tumor which was histologically a DA, found to be positive BRAF p.V600E and negative for H3 K27M and CDKN2A deletion could be structured as follows:
The term “Diffuse low-grade glioma, MAPK-altered” was introduced in the WHO 2021 classification to clearly differentiate this neoplasm from adult-type diffuse gliomas. 6 This entity does not currently have an assigned WHO grade. The integration of histology and molecular information into a single report allows for a more complete classification of pLGG and provides essential information needed to guide clinical decision making, including risk-stratification and selection of therapies.
Pediatric High-Grade Gliomas (pHGG)
pHGG are less common than pLGG, accounting for approximately 16% of brain tumors in children. 7 , 8 Histologically, this encompasses morphologies previously given WHO grades 3 and 4, namely anaplastic astrocytomas (AA) and glioblastomas (GBM), respectively, as well as anaplastic PXA (WHO grade 3) and anaplastic oligodendrogliomas (WHO grade 3). These tumors are often infiltrative on both imaging and histology. Examples of pHGG histologies are presented in Figure 3. Like their low-grade counterparts, pHGG can be found throughout the neuroaxis and thorough classification requires the integration of radiologic, histologic and molecular features.
Morphologies Encountered in Pediatric-Type High Grade Gliomas
pHGG are typically diffusely infiltrating gliomas (most frequently astrocytic in nature) which demonstrate mitotic activity, increased cellularity and nuclear atypia. In addition, these tumors may contain necrosis (which can be palisading) and/or microvascular proliferation. A subset of pHGG may have a primitive/embryonal appearance, a finding which is associated with specific molecular alterations (H3.3 p.G34R/V and MYCN amplifications) as discussed below. Loss of nuclear OLIG2 staining may be present in these primitive-appearing tumors (Figure 3(B)) though most demonstrate positive staining for GFAP, aiding in proper classification. Though nuclear atypia is a common feature of pHGG, significant nuclear pleomorphism may be encountered in the setting of cancer predisposition syndromes such as Li-Fraumeni (Figure 3(H)) and mismatch repair deficiency (MMRD) syndrome. Nuclear pleomorphism may also raise the possibility of an anaplastic PXA, especially when other histologic features are present (e.x. xanthomatous cells). Occasionally, some pHGG may not fit into a well-defined WHO category. Such tumors may be microscopically and radiographically well-circumscribed but have overt high-grade histology and/or lack astrocytic differentiation. For these instances, a diagnosis of pHGG, NOS or pHGG, NEC may be warranted as discussed above. 9
Molecular Findings in pHGG
Mutations in histone genes are the most common molecular alterations in pHGG (seen in approximately half of these neoplasms), and different histone mutations are associated with tumors of the cerebral hemispheres versus midline gliomas. The H3.3 p.G34R/V mutation is almost exclusively seen within tumors of the cerebral hemispheres, most commonly in the adolescent and young adult populations. 46 The term “diffuse hemispheric glioma, H3 G34-mutant” has been given to this entity in the WHO 2021 classification. 6 Approximately 25% of these tumors have primitive/embryonal morphology. H3 G34-mutant pHGG also commonly have co-occurring mutations in ATRX and TP53; mutations in IDH1/2 and H3 p.K27M are not seen in these gliomas. 46 , 47 H3 G34-mutant tumors correspond to WHO grade 4. 2 , 6 Though they are associated with somewhat longer survivals than adult-type IDH-wildtype GBM, the reported median survival is only 18.0 months, and the two-year overall survival is 27.3%. 46 , 47 In one study, H3 G34-mutant tumors were found to have poor outcomes similar to those of H3 K27M-mutant tumors. 48
In contrast to the hemispheric location associated with the H3.3 p.G34R/V mutation, H3 K27M-mutant tumors (comprises both H3.3 p.K27M and H3.1 p.K27M mutations) are mostly found within the midline. Between 63–95% of diffuse intrinsic pontine gliomas (DIPG) are positive for this mutation as are up to 50-64% of infiltrating tumors of other midline locations including the thalamus and spinal cord. 46 , 49 The 2021 WHO classification has been updated to allow for any diffuse midline glioma with loss of H3K27me3 to be given the designation “diffuse midline glioma, H3 K27-altered”. This includes infiltrative, astrocytic tumors of the midline with either H3.3, H3.2 or H3.1 p.K27M/I mutation, as well as tumors without H3 K27 mutations but with EZHIP over-expression or EGFR mutations. Diffuse midline glioma, H3 K27-altered corresponds to WHO grade 4. As discussed above, the presence of the H3 K27M mutation in tumors outside the midline or in non-infiltrative neoplasms does not warrant this diagnosis or automatic grade 4 designation, 43 though these tumors still tend to have poor outcomes. The majority of diffuse midline gliomas histologically AA or GBM. These tumors often present at a younger age than H3 G34-mutant tumors, especially those located in the pons where the median age at presentation is five years. 46 , 49 Co-occurring mutations are common in H3 K27M-mutant tumors including TP53 (60%) and ATRX (15%); mutations in BRAF and PDGFRA as well as several others have also been described. 46 H3.3 p.K27M and H3.1 p.K27M mutations are almost always mutually exclusive with H3.3 p.G34R/V and IDH1/2 mutations. H3 K27M-mutant pHGG have significantly shorter survivals than do H3 K27M-wildtype midline tumors with average overall survival of around one year. 30 , 50
As with pLGG, IDH mutations are also present in a small subset (∼6%) of pHGG, typically presenting in adolescents and young adults. 46 Mutations in TP53 and ATRX commonly co-occur in IDH-mutant astrocytomas, and 1p/19q-codeletions are seen in adult-type oligodendrogliomas. For these adult-type, IDH-mutant gliomas, new nomenclature was proposed 2 and adopted in the WHO 2021 classification. 6
Approximately half of pHGG fall into the category of “H3-/IDH-wildtype. 46 , 51 Of this group, a subset harbors the BRAF p.V600E SNV, often with co-occurring deletions in CDKN2A/CDK2NB. 46 , 52 These tumors have may have epithelioid histologies, including anaplastic PXA and epithelioid GBM (Figure 3(G)). Amongst pHGG, those with BRAF p.V600E SNV have better prognoses (2 year survival of 67%). 46
A subset of H3-/IDH-wildtype pHGG have chromosome 2 gains along with amplifications in genes such as MYCN, EGFR, and CDK6. These tumors with MYCN amplifications often have primitive/embryonal histology 51 and may show large cell anaplasia with prominent nucleoli as seen in medulloblastoma. These tumors are associated with poor outcomes and have a median overall survival of 14 months. 46
Finally, a subset of pHGG are found in the setting of cancer predisposition syndromes including Li-Fraumeni (due to germline mutations in TP53) and biallelic (constitutional) mismatch repair deficiency syndrome (bMMRD, caused by biallelic mismatch repair gene variants, PMS2 being the most common) or Lynch syndrome (mono-allelic germline mutations in mismatch repair genes). bMMRD-associated tumors may morphologically demonstrate marked pleomorphism. For patients with multiple childhood tumors, a family history of cancers (particularly of the gastrointestinal tract) and tumors with numerous molecular alterations, such syndromes should be considered. Additionally, bMMRD has significant clinical overlap with neurofibromatosis type 1 including café au lait macules, and parental consanguinity is seen in half of cases. 53
An Approach to Molecular Testing
Similar to pLGG, molecular findings provide important information for classification and risk stratification of pHGG. A single-method approach with an NGS panel which detects alterations seen in pHGG can be employed if available; however, like pLGG, step-wise approaches using IHC, FISH, PCR, and SNP arrays can be applied to pHGG. Figure 4 illustrates one such approach. Tumor location factors into this scheme; if the tumor is in a midline location, IHC for H3 K27M and H3K27me3 can be used to identify H3 K27-altered tumors. The H3 K27M mutation can also be detected by PCR. For hemispheric tumors, immunostains for H3.3 p.G34R and H3.3 p.G34V are available; however sequencing is more specific. 54
When mutation-specific immunostains are not available, IHC for OLIG2, p53, and ATRX can also be helpful in raising the suspicion for H3 G34-mutant hemispheric tumors as these gliomas often harbor co-occurring alterations in TP53 and ATRX and are negative for OLIG2. For hemispheric gliomas which are negative for H3.3 p.G34R/V and for all midline tumors (regardless of H3 K27M status), IHC for BRAF p.V600E may be done.
In older children, IDH1 p.R132H IHC should be performed. If positive, strong, diffuse staining or a total absence of staining for p53 as well as loss of nuclear ATRX staining supports the diagnosis of an IDH-mutant astrocytoma, and testing to confirm 1p/19q-codeletion is needed for a diagnosis of adult-type oligodendroglioma.
For pHGG which fall into the H3-/IDH-wildtype category, staining to detect other possible driver mutations (p53, BRAF p.V600E, and mismatch repair proteins) can be attempted. As IHC for p53 can often be difficult to interpret, additional molecular testing (such as NGS or PCR) to detect/confirm mutations in TP53 may be needed in some cases. In tumors with positive BRAF p.V600E staining, FISH or SNP array can be used to assess for CDKN2A deletions. FISH is also useful in detecting MYCN amplifications. In pHGG where this approach does not identify a driver mutation, SNP array, methylation array or NGS may then be considered.
As with pLGG, a diagnosis which integrates histologic and molecular information can be given in a layered format for pHGG. An example report for a midline GBM found to be a H3 K27-mutant follows:
Infantile Gliomas
A unique subgroup of pediatric gliomas is those occurring in infants under one year of age, referred to as infantile gliomas. Infantile gliomas can be histologically low- or high-grade. PA, gangliogliomas, desmoplastic infantile astrocytoma/gangliogliomas, DA, AA, and GBM may all present in infancy. 55 Additionally, both low- and high-grade tumors which are difficult to histologically classify are commonly seen in this age group. 55
Molecular testing of infantile gliomas has led to the discovery that some infantile CNS tumors, particularly the high-grade gliomas, harbor fusions not seen in gliomas of older children and adults. In a large study of 150 patients with infantile gliomas, over one third of these tumors were found to have receptor tyrosine kinase (RTK) alterations, specifically fusions in ALK, ROS1, NTRK, and MET genes. 56 These RTK-driven tumors were located in the cerebral hemispheres and were comprised of both high- and low- grade histologies though they are most frequently seen in high-grade tumors. In fact, 83% of infantile high-grade gliomas in this study had fusions in either the ALK, ROS1, NTRK, or MET genes. Many infantile low-grade gliomas, particularly tumors of the midline, were found to have RAS/MAPK pathway alterations. 56 This included BRAF fusions, BRAF p.V600E SNV and FGFR1 fusions. In contrast, RAS/MAPK pathway alterations were not found in infantile high-grade gliomas. Additionally, alterations associated with pHGG in older children such as H3 K27M, H3.3 p.G34R/V and IDH1 p.R132H were not seen in infantile high-grade gliomas. 56 These molecular findings indicate that infantile high-grade gliomas are a unique subset of pediatric gliomas. They are now included as a new tumor entity in the WHO 2021 classification (termed “infant-type hemispheric glioma”) and fall under the umbrella of pediatric-type diffuse high-grade gliomas.
The behavior of these infantile gliomas also differs from gliomas in older children and adolescents, and histologic grading may not be as predictable of outcome. Specifically, in comparison to gliomas of children ages 1–18 years, infantile high-grade gliomas have better outcomes while infantile low-grade gliomas exhibit more aggressive behavior. 56 Survival may be related to molecular findings and tumor location: hemispheric tumors with ALK, ROS1, NTRK, and MET fusions have an intermediate clinical outcome, hemispheric low-grade gliomas with RAS/MAPK-pathway alterations have excellent long-term survival, and midline RAS/MAPK-driven tumors have relatively poor outcomes. 56 The location of infantile low-grade gliomas with RAS/MAPK-pathway alterations (midline versus hemispheric) likely contributes to differences in outcome; however, these midline tumors, especially those located in the optic pathway/hypothalamus, often progressed and had significantly lower overall survival rates than optic pathway/hypothalamic gliomas in older children. 56
As with tumors of older children, infantile gliomas may benefit from treatment with targeted therapies. In addition to therapies aimed at the RAS/MAPK-pathway for infantile low-grade gliomas, RTK-driven tumors may also benefit from targeted therapies, and clinical trials are ongoing. As these tumors represent a unique subgroup of pediatric gliomas, additional studies will be necessary to further characterize infantile gliomas and better understand how histology, location and molecular alterations may predict outcomes.
For gliomas in infants, the above proposed schemes for molecular testing may be attempted, particularly for those which are histologically low-grade (Figure 2). As most of the high-grade tumors fall into the category of H3-/IDH-wildtype, testing for ALK, ROS1, NTRK, and MET fusions should be considered (Figure 4). IHC and/or FISH can be used to detect many of these alterations see in high-grade infantile gliomas. Alternatively, a NGS panel which includes the above genes may also be employed.
Conclusions
Here we have described the varied histologies and molecular findings of pLGG, pHGG, and infantile gliomas and presented possible approaches to identify common molecular alterations. pLGG are characterized by alterations causing upregulation of the RAS/MAPK pathway whereas pHGG commonly harbor mutations in histone genes. Infantile high-grade gliomas commonly have RTK alterations. As molecular testing becomes an integral part of the classification of brain tumors 1 , 6 as well as for selection of targeted therapies, either stepwise or single-method approaches to identifying clinically significant alterations will be necessary. By integrating histology (tumor type and histologic grade) with clinical information and molecular findings, the pediatric pathologist can more accurately classify pediatric gliomas. This classification can be used to better predict prognosis and identify possible therapies, both essential to clinical decision making.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.