
Abstract
Unique dental conditions in children include odontogenic cysts and tumors, hereditary dental diseases, developmental anomalies, and lesions associated with the eruption of the primary or permanent teeth. Many of these conditions have long lasting effects on the adult dentition in terms of affecting esthetics, function, and overall quality of life. Inherited dental syndromes affect the dental hard tissues specifically the enamel, dentin, and/or cementum in a generalized manner, involving both primary and permanent teeth. These conditions manifest in altered quality or quantity of the hard tissues, leading to fragility, tooth loss and dental diseases such as caries, periapical pathology, and periodontal disease. This category includes amelogenesis imperfecta, dentinogenesis imperfecta, dentin dysplasia, hypophosphatasia, and hypophosphatemia. Developmental defects such as regional odontodysplasia are defined by involvement of the primary and permanent dentition in a localized manner, identified in early childhood. This review will elaborate on the histologic findings in these selected dental conditions with a discussion on clinical and radiographic findings, as well as molecular features wherever appropriate.
Keywords
Introduction
Dental anomalies occur at the time of tooth development and manifest in qualitative, quantitative, and morphologic changes. Clinical identification of many of these anomalies occur at the time of eruption in childhood. The four stages of tooth development are (1) Bud stage, (2) Cap stage, (3) Bell stage, and (4) Late Bell stage. 1 Disruption of tooth development during these stages may result in anomalies of tooth number, shape or morphology, eruption pattern, and/or defects in enamel, dentin, and cementum. Tooth mineralization begins at the Bell stage. 1 The principal cells are ameloblasts, odontoblasts, and cementoblasts. Chronologic environmental insults, hormonal imbalances, or genetic mutations coding for critical proteins or enzymes will result in disruption or deficient matrix production and mineralization.
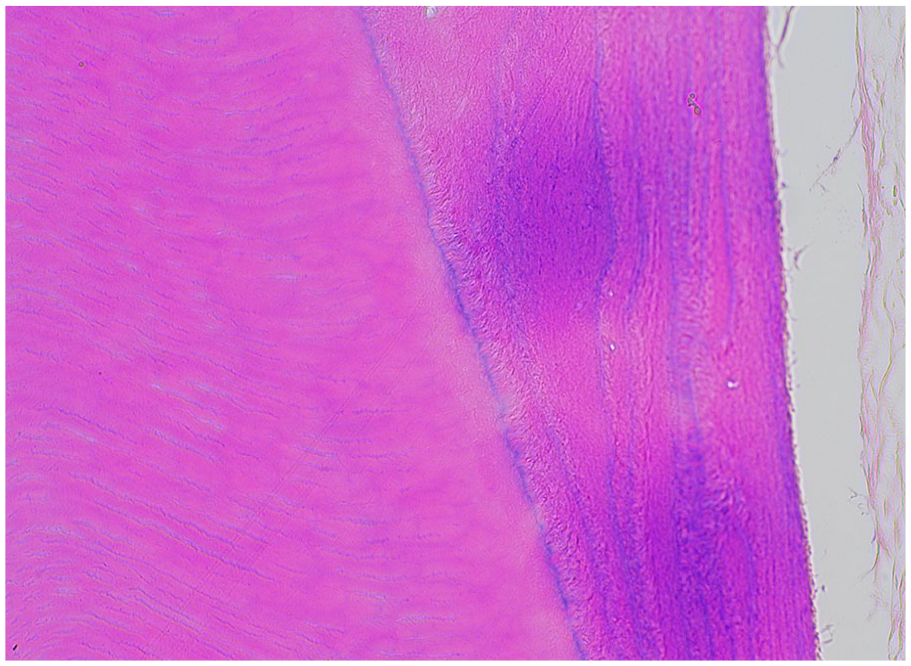
Dentin production or dentinogenesis is completed by odontoblasts. The odontoblasts line the primitive pulp, laying down the predentin which acts as a buffer zone.2,3 Odontoblasts secrete type 1 collagen, calcium ions, and proteins such as dentin sialophosphoprotein. 2 As dentin is deposited and mineralized, the odontoblasts retreat toward the pulp. The cytoplasmic processes elongate and remain encased in dentin tubules (Figure 1). 2 Dentin is approximately 70% mineral, 20% organic matrix, and 10% water. 4 Histologic examination of dentin after routine decalcification preparation reveals homogenous, eosinophilic mineralized hard tissue with linear tubules spanning the width of the dentin (Figure 2). Mutations in genes coding for dentin sialophosphoprotein and type 1 collagen result in disorganized dentin, as observed with dentinogenesis imperfecta and osteogenesis imperfecta type 1A, respectively.
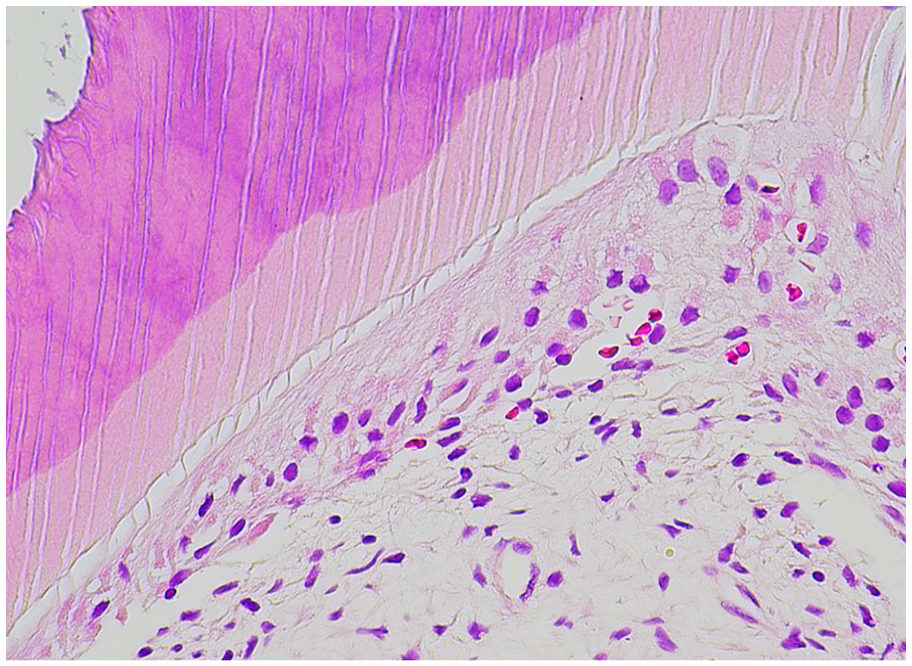
Odontoblasts. Odontoblast processes extend from the pulpal tissues into the dentin tubules.
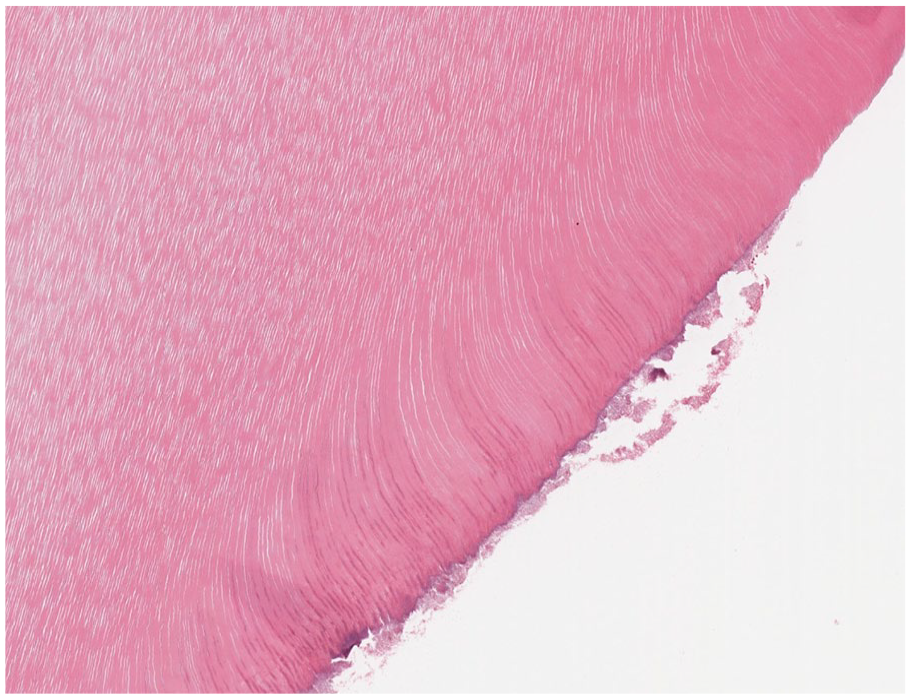
Dentin. Decalcified dentin demonstrates linear dentinal tubules that radiate from the pulpal tissues to the cementum or enamel interface.
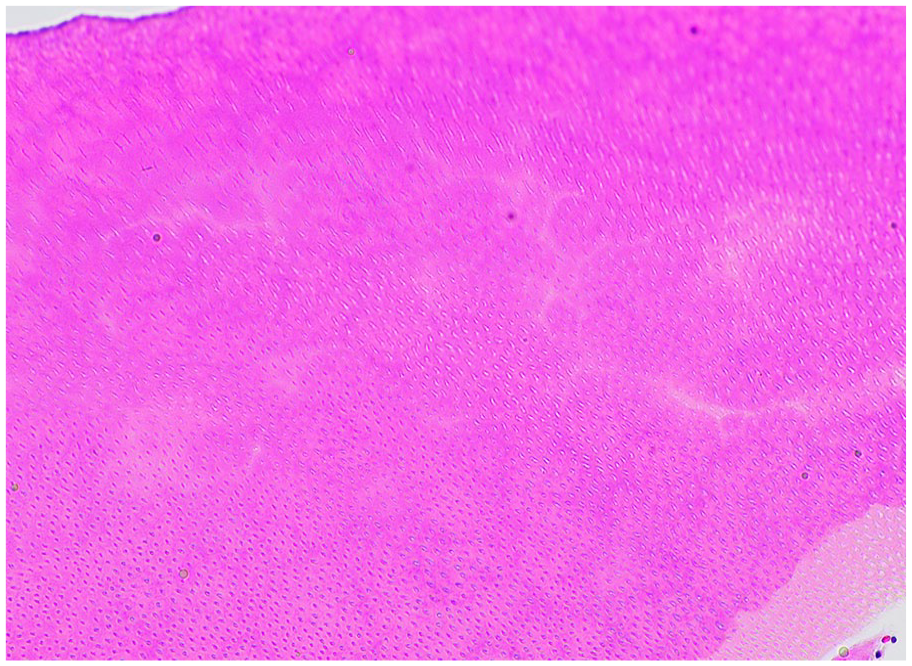
Enamel production and mineralization (amelogenesis) is driven by ameloblasts within the developing tooth bud and is completed by a series of consecutive stages: presecretory, secretory, transition, and maturation, ending with apoptosis of the ameloblasts and eruption into the oral cavity.5,6 Presecretory ameloblasts initiate the deposition of enamel matrix on the developing dentin and differentiate into secretory ameloblasts. During the secretory phase, ameloblasts deposit enamelin (coded by the ENAM gene), amelogenin (AMELX gene), and ameloblastin (ABN gene) along with matrix proteinase-20 (MMP-20 gene) into the matrix. 6 The high-protein enamel matrix is soft and cheesy during this stage.5-7 KLK-4 the transition and maturation stages is characterized by the activity of kallikrein-related peptidase 4 (gene) to remove the deposited proteins, converting enamel matrix to mature enamel which is composed of 96% hydroxyapatite and 4% protein.3,6,7 This high mineral to protein ratio explains the loss of enamel during the decalcification process, which is identified as an empty space on histologic examination. 3 Enamel matrix is observed in developing teeth or odontomas and presents as a basophilic, unmineralized material with a wavy, ruffled, “fish scale” pattern (Figure 3). Genetic defects in these proteins have been recognized in some forms of amelogenesis imperfecta.
Enamel. Enamel matrix (right), demonstrating a wavy “fish scale” appearance, juxtaposed to dentin (left), demonstrating dentinal tubules.
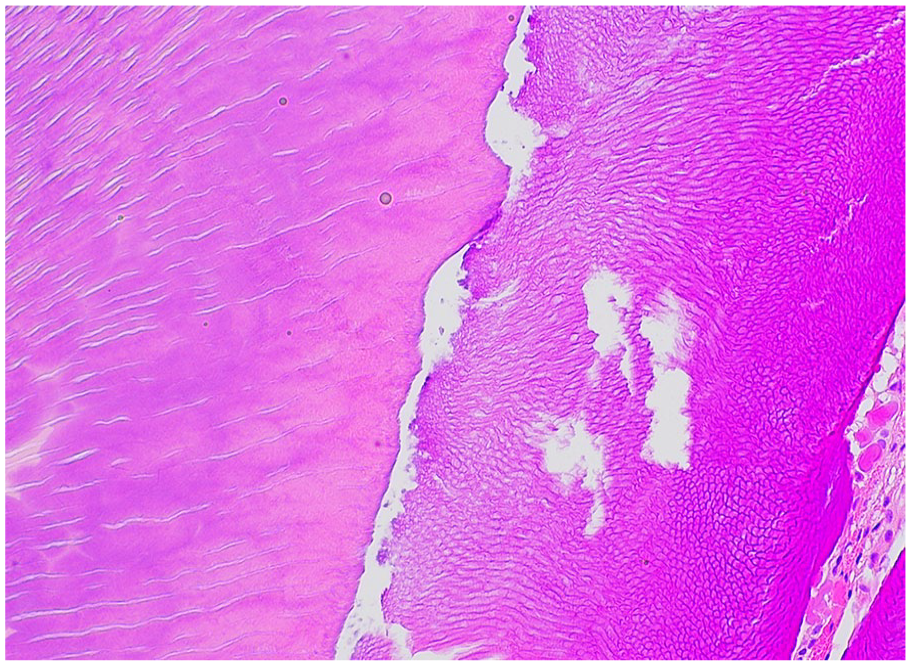
Cementogenesis is driven by cementoblasts, which are differentiated from cells in the dental follicle upon interaction with the root dentin. 1 The cementoblasts deposit cementum matrix on the dentin.1,3 Fibroblasts in the dental follicle simultaneously secrete collagen which insert in the mineralized cementum. 1 These collagen fibers insert into the alveolar bone, forming the principal fibers of the periodontal ligament, or Sharpey’s fibers, 1 Cementum is 45% to 50% inorganic hydroxyapatite and 50% organic material and water. Decalcification and histologic examination will reveal an acellular, laminated, mineralized material with histologic similarities to bone (Figure 4). Deficiencies in cementum production is seen in hypophosphatasia, resulting from abnormal tissue nonspecific alkaline phosphatase (TNSALP), which is widely expressed in the periodontal ligament cells, cementoblasts as well as odontoblasts. 8
Cementum. Cementum is characterized by laminated acellular mineralized material with scanty nuclei embedded (right), and is intimately associated with dentin (left).
Clinical mimickers of hereditary dental diseases include environmental insults at the time of tooth development, including fluorosis, chronologic enamel hypoplasia, molar-incisor malformation and intrinsic staining such as tetracycline staining. Environmental injuries affect the specific teeth developing at the time of the insult, whereas an inherited dental disease or syndrome will affect all the teeth in the primary and permanent dentition. The clinical and radiographic findings will assist in determining a localized or generalized process.
This review will examine the histologic and diagnostic features of some common and uncommon dental anomalies occurring in the pediatric population.
Inherited Dental Conditions
Amelogenesis Imperfecta (AI)
Clinical presentation
Amelogenesis imperfecta is a complex hereditary disorder of enamel matrix production, calcification and/or maturation, manifesting in enamel loss, fragility, and hypoplasia. AI is characterized by generalized involvement of the primary and permanent dentition, presenting with variability in enamel production, color, hardness, and surface texture. 9 AI is a clinical and radiographic diagnosis. Genetic testing is currently available for research purposes to identify pathogenic mutations. As more mutations are identified, the classification schemata will continue to evolve. Currently the recommended classification combines phenotype and mode of inheritance. 10 The phenotypes of AI include hypocalcified, hypoplastic, and hypomaturation (Figure 5). 9 These teeth may present with white-yellow-brown discoloration, ena-mel pitting or grooves, thin translucent enamel, or with cheesy soft enamel with rapid loss pre- and post-eruption. 9 The variable presentations are based on the type of enamel defect. AI can be isolated or associated with syndromes or other conditions including tricho-dento-osseous syndrome, nephrocalcinosis, cone rod dystrophy, and Kohlschutter syndrome. 11 A renal examination may be recommended upon diagnosing AI to rule out nephrocalcinosis. 9
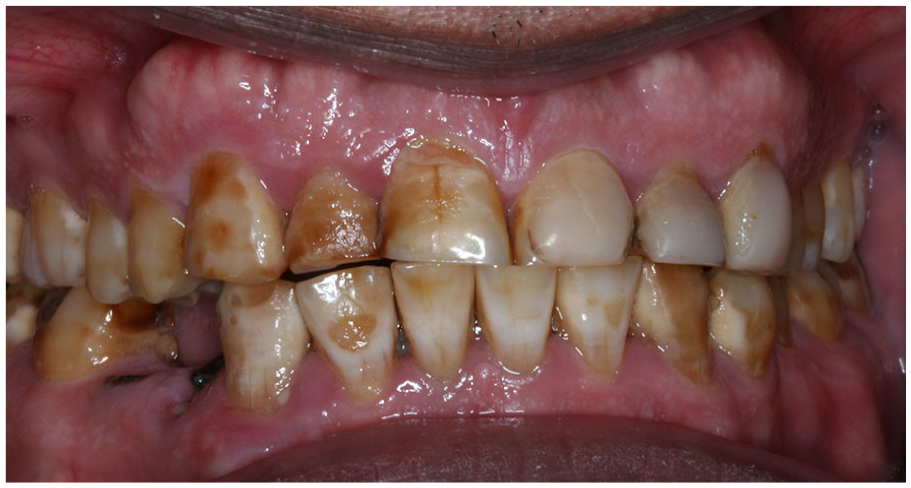
Amelogenesis imperfecta. Generalized involvement of all permanent dentition, characterized by mottled, discolored, pitted enamel. Marked attrition of the incisors is noted. (Courtesy of Dr. Elizabeth Bilodeau, University of Pittsburgh, used with permission).
Radiographic features
The teeth may exhibit thin enamel or significant enamel resorption pre- and post-eruption.
Gross features
The teeth may be malformed, have short clinical crowns and/or exhibit discoloration with defects on the enamel surface. Unaffected root cementum and dentin is a consistent feature.
Histologic features
Decalcification of these teeth will result in enamel loss due to the high content of hydroxyapatite. Decalcification is not recommended when examining teeth suspected to be affected by enamel dysplasias. The dentin, pulp, and cementum will not exhibit pathologic changes (Figure 6). The diagnosis is primarily made on clinical and radiographic findings.
Amelogenesis imperfecta. Decalcified tooth demonstrating intact dentin, with missing enamel. Histologic examination of a tooth affected by enamel conditions is not recommended. (Courtesy of Dr. Elizabeth Bilodeau, University of Pittsburgh, used with permission).
Molecular features
X-linked dominant, autosomal dominant, and autosomal recessive inheritance patterns have been identified in patients with amelogenesis imperfecta.9,12 Mutations in genes involved in enamel production include ENAM, AMELX, FAM83H, DLX3, MMP20, and KLK4.9,11 Sporadic cases have been reported. 11
Management
Patients will require long term treatment for caries and significant occlusal wear or attrition as well as esthetic rehabilitation. Full coverage restorations are often indicated, starting with the primary dentition. 13 The poor quality or quantity of enamel renders typical restorative bonding to be less effective. 13 The psychosocial impact of the altered esthetics and altered quality of life cannot be understated.
Dentinogenesis Imperfecta (DGI)
Other names: Hereditary opalescent dentin, Capdepont’s teeth.
Clinical presentation
This condition is an autosomal dominant disorder affecting both primary and permanent dentition, resulting from mutations in the DSPP (dentin sialophosphoprotein) gene which codes for multiple proteins involved in the mineralization of dentin. 14 Affected teeth present with a translucent, amber, or bluish color (Figure 7).9,14 Significant enamel fracture and attrition or wear occur soon after eruption.9,14 Dentin dysplasia type II, previously considered to be a separate condition, is now considered to be a more mild form of DGI. The reclassification of DGI and related conditions is further elaborated in Table 1.14,15 Patients with osteogenesis imperfecta type 1A with mutations in the COL1A1 gene may manifest identical dental changes to DGI.9,16 Other syndromes with clinical and radiographic presentations similar to DGI include Goldblatt syndrome, Schimke immune-osseous dysplasia, and Ehlers-Danlos type II. 16 Progressive sensorineural hearing loss has been reported in several families affected by DGI. 9
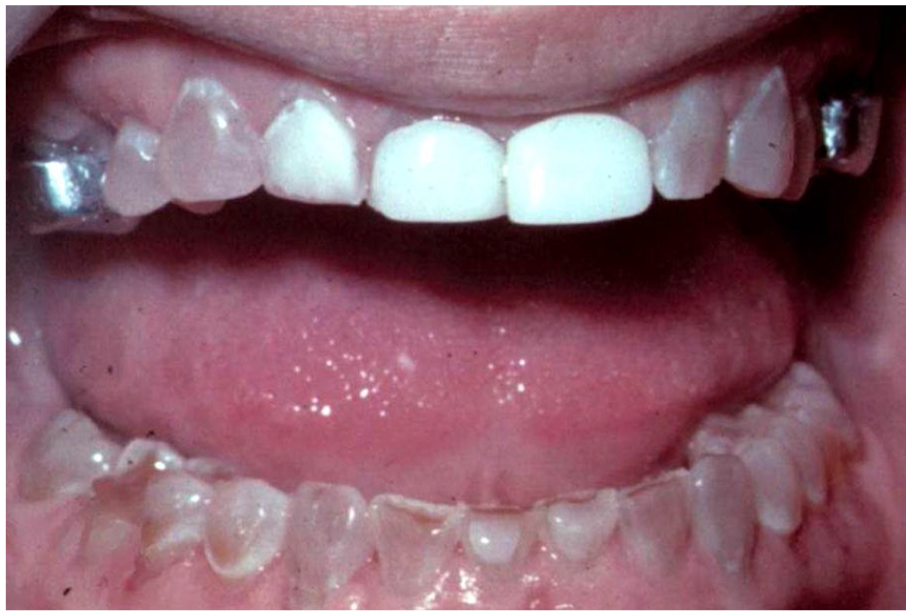
Dentinogenesis imperfecta. Generalized involvement of all primary dentition, characterized by bluish-amber coloration, marked attrition, and full coverage restorations of some of the dentition. (Courtesy of Dr. Elizabeth Bilodeau, University of Pittsburgh, used with permission).
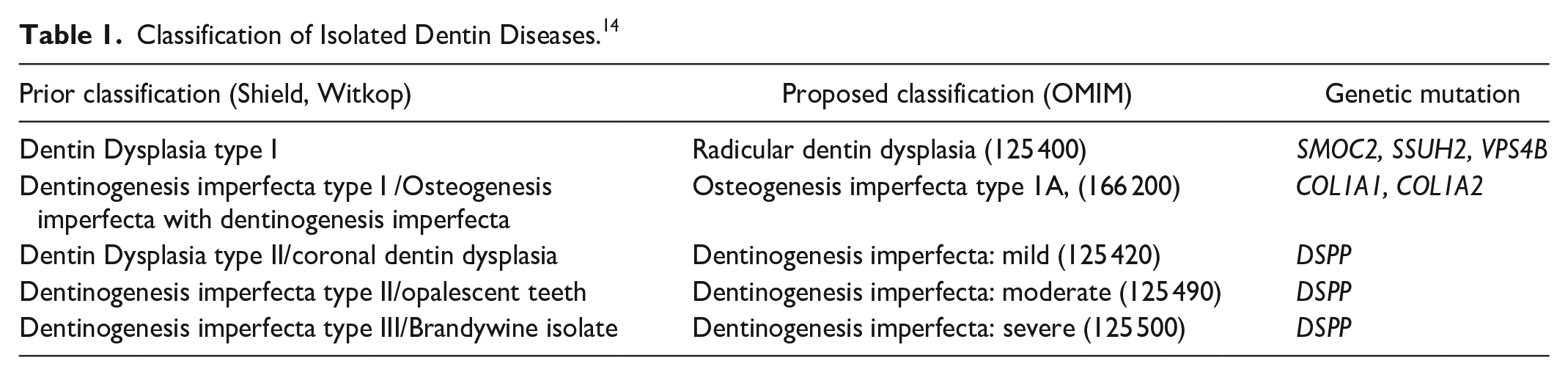
Classification of Isolated Dentin Diseases. 14
Radiographic features
Affected teeth demonstrate obliterated pulp chambers with bulbous crowns and cervical constriction, or a narrow crown-root junction (Figure 8). 9 The roots are spindly and thin (Figure 9). Periapical pathology is common due to pulpal necrosis from attrition and wear. The permanent dentition in some patients with mild DGI (Dentin dysplasia type II) demonstrate “thistle-shaped” or enlarged, ovoid pulp chambers. 9 The more severe manifestation of DGI may demonstrate massive pulps with thin enamel and dentin, which are called “shell teeth.” 9
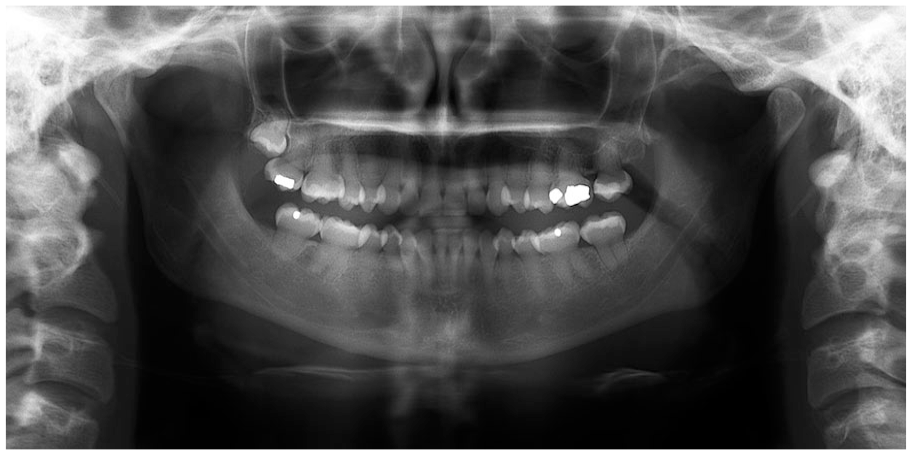
Dentinogenesis imperfecta. Panoramic radiographic demonstrating cervical constriction, pulpal obliteration, and spindled, thin roots.
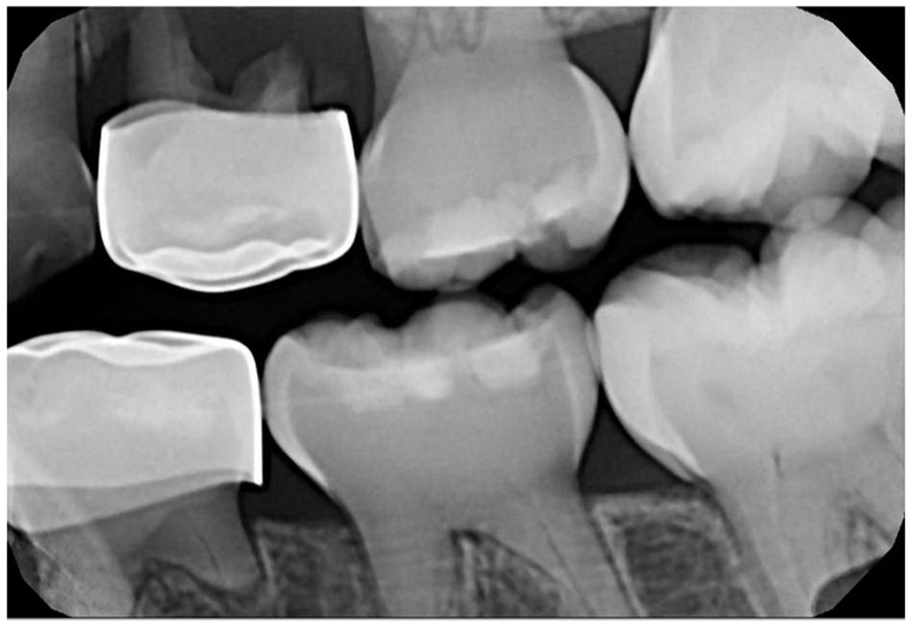
Dentinogenesis imperfecta. Bitewing radiograph demonstrating cervical constriction, pulpal obliteration, and spindled, thin roots. Periapical pathology involving tooth #I (left maxillary first primary molar) is noted.
Gross features
The teeth are bluish to amber colored, with short thin roots and a bulbous crown morphology. The enamel may be fractured or missing.
Histologic features
The characteristic features of DGI include abnormal, disorganized dentin with short tubules in a granular matrix (Figure 10). The dentin adjacent to the enamel is normal, however the circumpulpal dentin is affected. Interglobular calcifications can be seen with entrapped odontoblasts (Figure 11). 9 Pulp stones may be observed. 9 In patients with mild DGI, the root dentin is primarily affected.

Dentinogenesis imperfecta. Aberrant dentin with short, misshapen, irregular dentinal tubules, with scattered basophilic interglobular calcifications. (Courtesy of Dr. John Hellstein, University of Iowa, used with permission).

Dentinogenesis imperfecta. Interglobular calcifications. (Courtesy of Dr. John Hellstein, University of Iowa, used with permission).
Molecular features
Mutations of the DSPP gene with 38 pathogenic variants, with an autosomal dominant inheritance pattern, have been identified. 14 The Shield and Witkop classifications were historically used to separate out different dentin diseases, however the identification of common DSPP gene mutations suggests these conditions exist on a spectrum of severity, rather than represent separate conditions (Table 1).9,14,15
Management
Genetic confirmation may be indicated for diagnosis in equivocal cases. Protection of the existing dentition by full coverage restorations is often necessary, as these teeth are at risk for enamel fracture, attrition, and pulpal necrosis. These patients require routine, long term follow up with their dentists.
Dentin Dysplasia type 1 (DD-I)
Other names: Rootless teeth, radicular dentin dysplasia.
Clinical presentation
Dentin dysplasia type I (DD-I) is an uncommon autosomal dominant condition, characterized by significant root shortening or aplasia which may lead to tooth mobility, pulpal necrosis, and premature tooth exfoliation. 9 Both primary and permanent dentitions are affected. 9 The coronal enamel and dentin appear clinically normal. 9 Other conditions with similar morphologic and clinical features to DD-I include familial tumoral calcinosis, vitamin D-resistant rickets, a history of chemotherapy or radiation therapy to the jaws in childhood, and molar-incisor malformation (MIM). 9 MIM presents as morphologic changes to the roots of affected incisors, first permanent molars and occasionally canines, secondary to environmental or medical insults prenatally, or during the first year of life.17-20 Other teeth may rarely be affected. 17 The specific pattern of teeth affected and a positive history of a medical event supports the diagnosis of MIM over DD-I.
Radiographic features
Dental radiographic imaging is significant for generalized involvement of the teeth with minimal to absent tooth root development (Figure 12). The primary dentition exhibits more severe root malformations than the permanent dentition. The permanent dentition may exhibit more variability in root shape and length. 9 MIM must be considered in the differential diagnosis of radiographic radicular pathology. The molars are clinically normal, however the roots are spindly, malformed, and may be missing (Figure 13). The affected incisors and canines may exhibit malformed roots and a cervical wedge-shaped defect (Figure 14). 18 The remaining dentition is otherwise uninvolved.
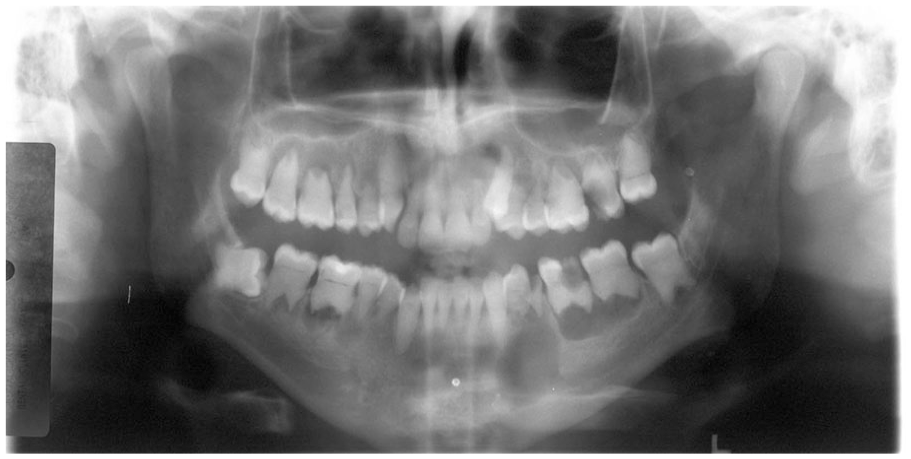
Dentin dysplasia type 1. Panoramic radiograph demonstrating generalized involvement of permanent dentition, exhibiting markedly shortened roots with multifocal areas of periapical pathology. (Courtesy of Dr. Elizabeth Bilodeau, University of Pittsburgh, used with permission).
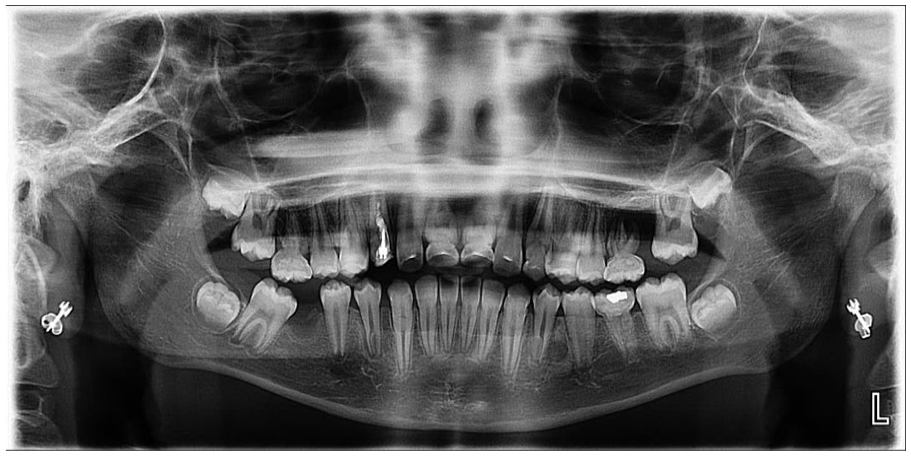
Molar-incisor malformation. Panoramic radiographic demonstrating involvement of the first permanent molars, maxillary canines, and maxillary central incisors. The affected dentition have abnormal spindled roots and cervical constriction. Tooth #30 (right mandibular first permanent molar) was extracted due to odontogenic infection.
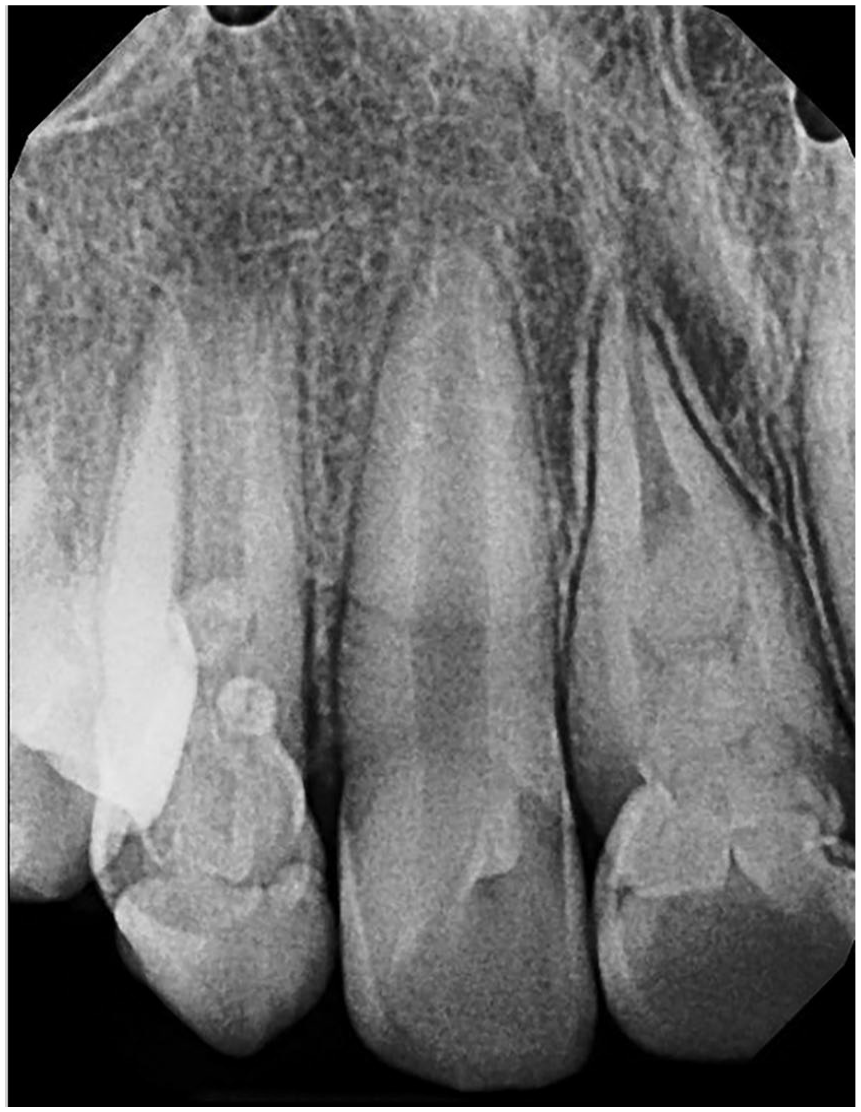
Molar-incisor malformation. Periapical radiograph demonstrating involvement of the right maxillary canine and right central incisor. The roots exhibit amorphous globular dentin. The central incisor demonstrates a wedge-shaped cervical defect.
Gross features
These teeth demonstrate normal crown morphology with significant root shortening or root aplasia.
Histologic features
The coronal enamel and the thin layer of dentin adjacent to the enamel are normal. The circumpulpal dentin demonstrates whorled disorganized dentin forming calcified masses obliterating the pulp chamber, resembling a “stream flowing around boulders.” (Figures 15 and 16).9,21 The dentin tubules are short, sparse, and fragmented (Figure 17). Abnormal globular radicular dentin may be observed in MIM as well. 17 Clinical and radiographic correlation is recommended to determine if the histology supports DD-I or MIM.
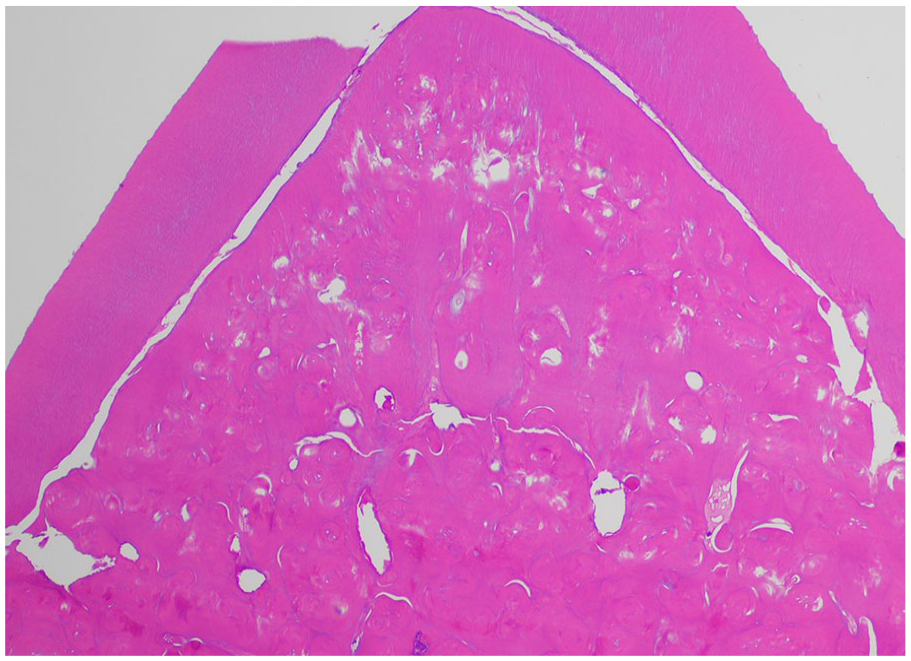
Dentin dysplasia type 1. “Normal” dentin is located at the periphery, with abnormal, whorled dentin occluding the pulp. (Courtesy of Dr. John Hellstein, University of Iowa, used with permission).
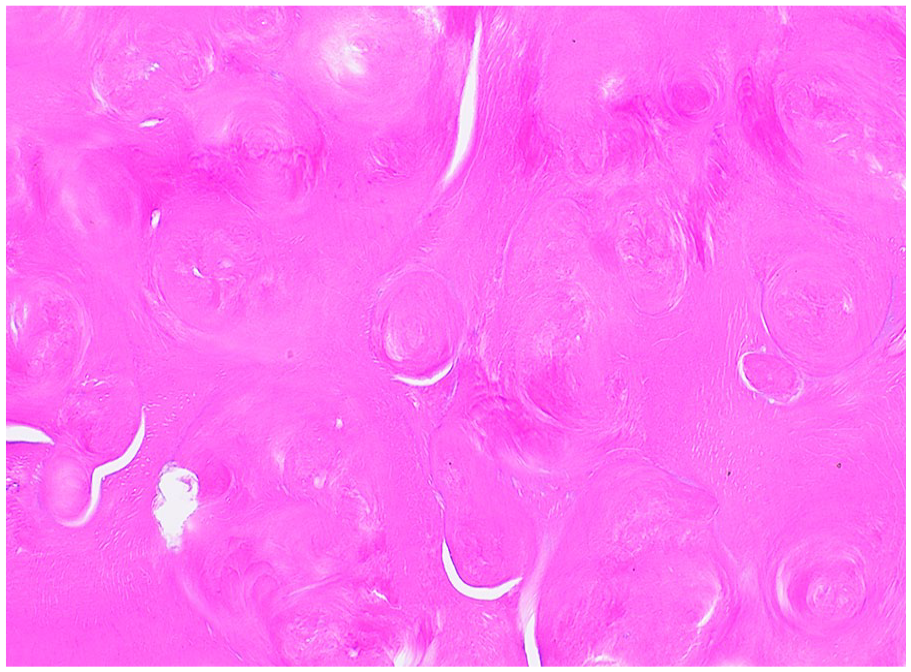
Dentin dysplasia type 1. Abnormal whorled dentin forms acellular masses. (Courtesy of Dr. John Hellstein, University of Iowa, used with permission).
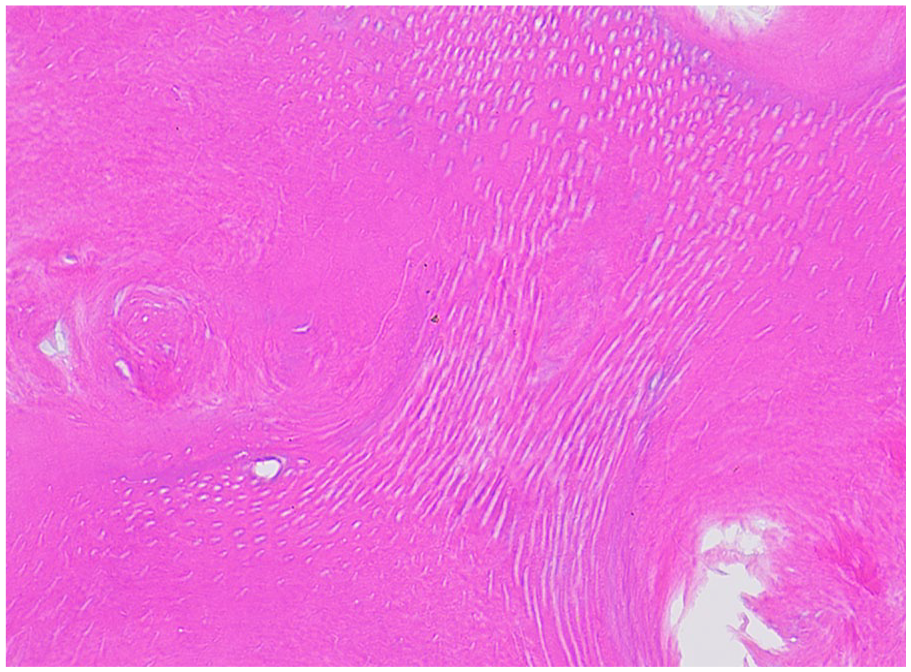
Dentin dysplasia type 1. Tubular dentin with short, misshapen tubules are identified. (Courtesy of Dr. John Hellstein, University of Iowa, used with permission).
Molecular features
DD-I demonstrates an autosomal dominance inheritance pattern with genetic heterogeneity. Multiple mutant genes in several families affected by DD-I have been identified including SSUH2, SMOC2, and VPS4B.22,23 Genetic testing is performed for research purposes, not for diagnostic assistance.
Management
Early dentition loss from lack of periodontal stability, and spontaneous tooth devitalization are common. 9 Prevention and maintenance of the existing dentition is key. Therapies are aimed toward addressing dental disease such as pulpal necrosis and subsequent inflammatory periapical disease as well as tooth replacement.
Hypophosphatasia (HPP)
Clinical presentation
Hypophosphatasia is a hereditary metabolic bone disease with deficiency of tissue nonspecific alkaline phosphatase (TNSALP). The characteristic clinical features include musculoskeletal anomalies similar to rickets and premature tooth exfoliation due to the decreased or lack of root cementum. There is quite a spectrum of symptoms ranging from severe which are lethal to mild forms which may have non-specific features.17,25 The most common presenting feature is premature tooth loss, usually prior to 3 years of age, of primary teeth with the root intact. Often the diagnosis is delayed due to clinicians not being aware of this entity.17 -19
Radiographic features
The radiographic features may be non-specific and are not pathognomonic. The height of the alveolar bone may be reduced. Widened pulp chambers, thin dentin, and abnormal roots have been described.17,19
Gross features
The exfoliated teeth will demonstrate an intact root (Figure 18).
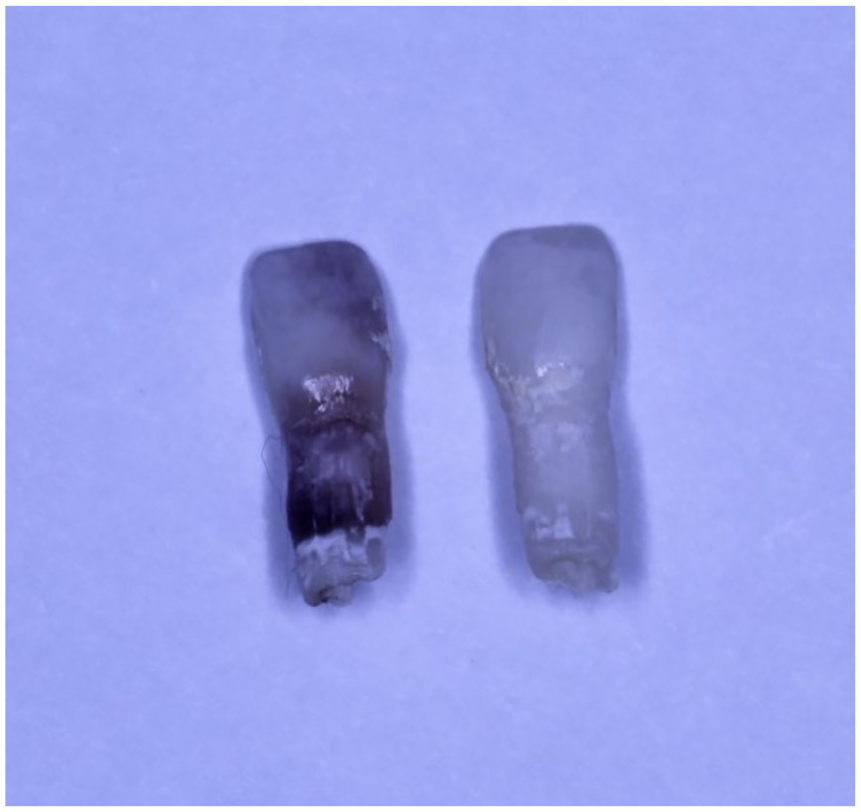
Hypophosphatasia. Gross photograph of prematurely exfoliated primary central incisors, with intact roots.
Histologic features
Teeth are rarely submitted for pathologic review. A definitive diagnosis of hypophosphatasia should not be made on histology alone. The cementum is abnormal and may be hypoplastic or completely absent (Figure 19). Changes in the dentin and enamel are not consistently reported.24,25-28
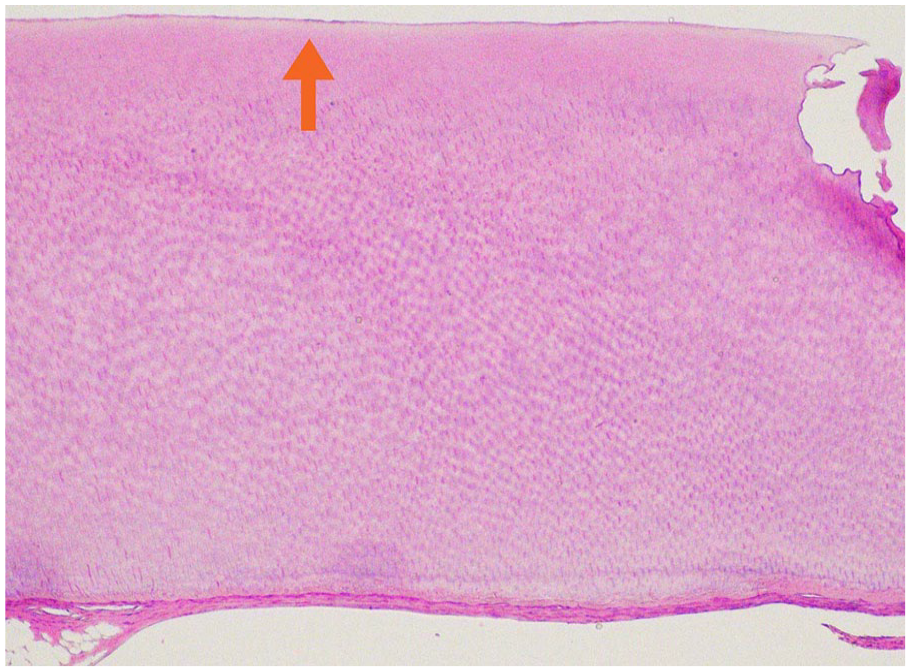
Hypophosphatasia. High power view of a root of an affected tooth, composed only dentin. Cementum is not identified. The arrow is pointing to a thin eosinophilic band at the outer surface of dentin.
Molecular features
HPP is a diagnosis now often made by genetic testing following clinical suspicion, although not necessary in straightforward cases. HPP is due to mutations in the gene ALPL on chromosome 1p36.1, which encodes the enzyme TNSALP. A deficiency in TNSALP enzyme activity leads to elevations in several TNSALP substrates, including inorganic pyrophosphate (PPi). Elevated extracellular levels of PPi inhibits bone mineralization which manifests as rickets and bone deformation in infants and children and as osteomalacia once growth plates close, along with muscle weakness. There is high allelic heterogeneity and can be inherited in an autosomal dominant or recessive pattern.17,29 -31
Management
Until recently, treatment was largely supportive for all types. TNSALP enzyme replacement therapy (asfotase alfa or Strensiq injections) has been approved for patients with perinatal/infantile and juvenile onset HPP and reduces the enzyme substrate levels. 31 The oral health, in addition to other symptoms such as skeletal gait and muscle pain, have shown to improve with enzyme replacement therapy.24,31,32
Vitamin D Deficiency Rickets, Vitamin-D Resistant Rickets
Other names: hypophosphatemia, hypophosphatemic rickets, X-linked rickets, hereditary hypophosphatemic rickets types I and II.
Clinical features
Rickets may be caused by a decrease in vitamin D (vitamin D deficiency rickets) or a genetic mutation in the vitamin D receptor (vitamin D resistant rickets). Systemic clinical features of both conditions include short stature, bowed or knock-kneed legs, and bone pain during physical activities (Figure 20).8,33 Dental manifestations in both conditions occur as a consequence of the hypercalcemia and hyperparathyroidism, secondary to decreased vitamin D. The parathyroid hormone 1 receptor (PTH1R) is expressed in osteoclasts as well as ameloblasts, odontoblasts, and cementoblasts and is sensitive to fluctuations in parathyroid hormone. 8 Enamel hypoplasia and fragility, as well as abnormal dentin and cementum deposition are observed, which lead to advanced periodontal disease, attrition, spontaneous pulpal necrosis, microabscesses, and premature tooth exfoliation.

Vitamin D Resistant Rickets. Affected individual demonstrating bowed legs. (Courtesy of Dr. Elizabeth Bilodeau, University of Pittsburgh, used with permission).
Radiographic features
The teeth may demonstrate radiographic or histologic evidence of high pulp horns approaching the dentin-enamel junction. Microscopic pulpal exposures lead to pulpal necrosis and subsequent periapical pathology (Figure 21). Involvement of multiple teeth that are otherwise intact (negative for caries or restorations) with spontaneous pulpal necrosis is suggestive of this condition.
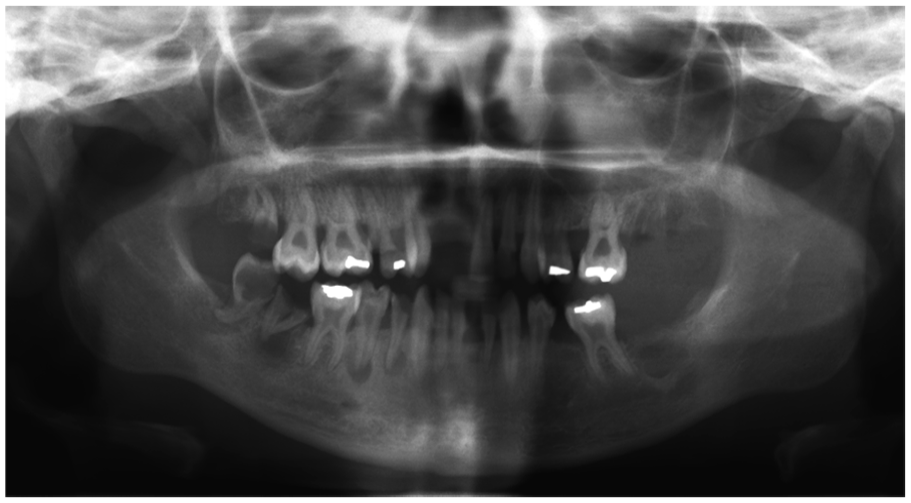
Vitamin D Resistant Rickets. Panoramic radiograph demonstrating multifocal periapical pathologies. (Courtesy of Dr. Elizabeth Bilodeau, University of Pittsburgh, used with permission).
Gross features
These teeth are morphologically intact.
Histologic features
Decalcified teeth will reveal necrotic pulp and high pulp horns, manifesting in clefts approaching the dentin-enamel junction. 9 The dentin demonstrates interglobular dentin with clefts in the enamel and dentin (Figure 22). 9 The pulp tissue is often nonvital. 9 The pathologist is more likely to receive the resulting periapical tissues which are non specific granulation tissue or inflamed odontogenic cysts. The diagnosis of hypophosphatemia cannot be made on histology alone.
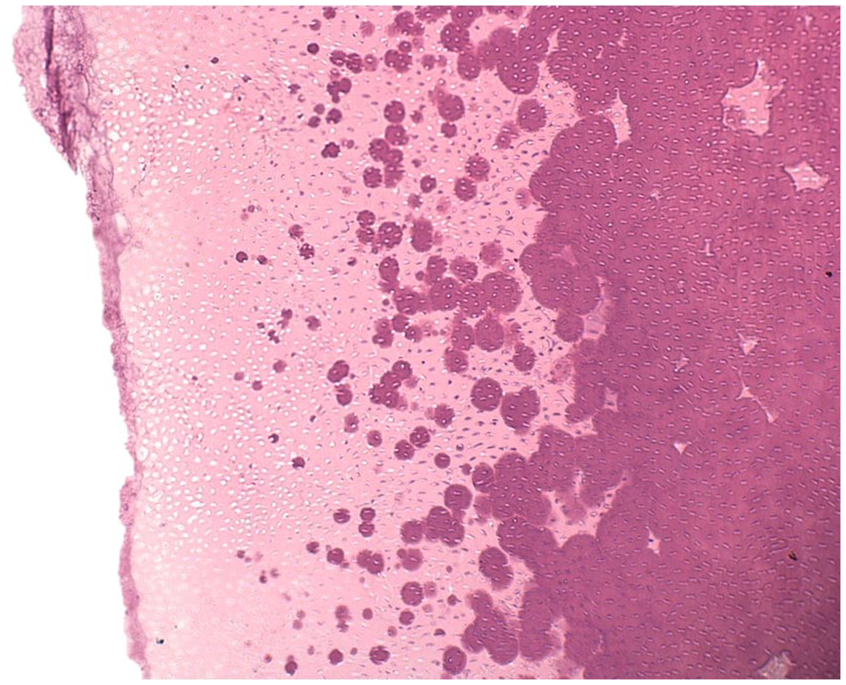
Vitamin D Resistant Rickets. Abnormal interglobular dentin. (Courtesy of Dr. Elizabeth Bilodeau, University of Pittsburgh, used with permission).
Molecular features
The inheritance pattern for hereditary vitamin D resistant rickets is most commonly X-linked dominant, although autosomal recessive and autosomal dominant patterns exist. 28 X-Linked hypophosphatemia is caused by a mutation in the PHEX gene (phosphate-regulating gene with endopeptidase activity on the X chromosome).8,9,33 Autosomal dominant hypophosphatemia is caused by a mutation in FGF23 gene (fibroblast growth factor 23).8,33 Vitamin D deficient rickets is a disease of nutritional deficiency and not related to genetic inheritance.
Management
Genetic testing is indicated to identify the causative mutation. Vitamin D deficient rickets is treated with vitamin D supplementation and hereditary Vitamin D resistant rickets is typically treated with supplemented calcitriol. Careful long term management of the existing dentition is required to prevent future pulpal necrosis and tooth loss. Prophylactic full coverage restoration and careful restorative techniques may be indicated to prevent microscopic exposure of the pulp tissues.
Developmental Tooth Anomalies
Regional Odontodysplasia: (ROD)
Other names: Ghost teeth.
Clinical presentation
This uncommon developmental condition is characterized by defects in enamel, dentin, and pulp tissue development. It affects both primary and permanent dentition in a segment of the jaws, most commonly the anterior maxilla. It is a non-inherited developmental tooth anomaly which can be isolated or associated with some syndromes and neural, vascular, or growth disorders. 9 The affected teeth may be small, brown, or yellow with malformed morphology.9,34 The teeth may exhibit delayed eruption or failure to erupt.9,34,35 Often the erupted teeth develop caries and inflammatory periapical pathology.9,21,34
Radiographic features
Characterized as “ghost teeth,” the teeth demonstrate abnormally thin enamel and dentin with expanded pulp tissues. 9 The hypoplastic dental tissues may create a “fuzzy” radiographic appearance, with indistinct borders (Figure 23).9,34 Periapical pathology may be observed.
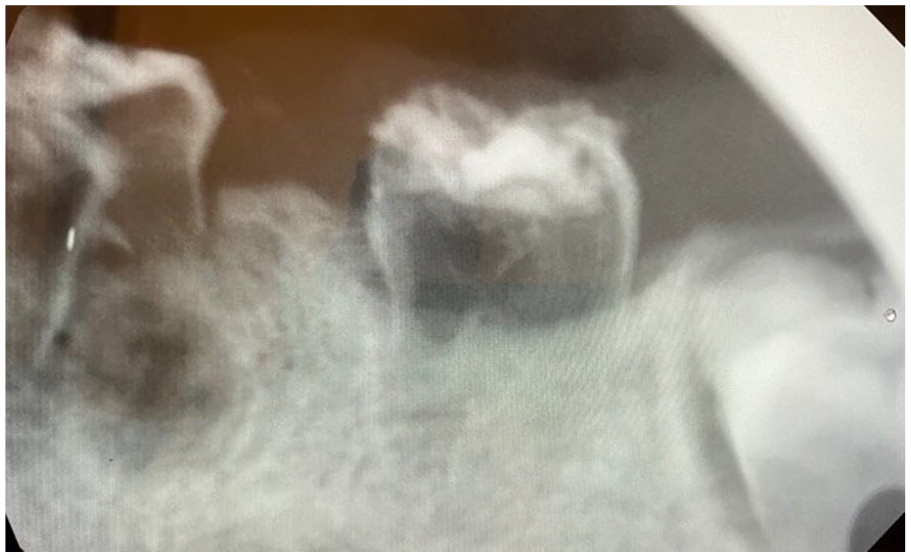
Regional odontodysplasia (ROD). Periapical radiograph demonstrating primary molars with fuzzy, indistinct enamel, enlarged pulps, and thin dentin. Periapical pathology is noted at the apex of tooth #L (left mandibular first primary molar). The developing permanent molar appears to be unaffected.
Gross features
The crowns are discolored and small, with grooves, pits and distorted morphology.35 The enamel is reported to be soft and easily punctured with a probe. 9 The roots are abnormally shaped and hypoplastic.34,35
Histologic features
The enamel is irregular and hypoplastic with a laminated appearance and residual enamel matrix.9,35 The dentin is disorganized, with clefts scattered throughout areas of amorphous and interglobular dentin and shortened, disorganized dentinal tubules.9,34,35 The pulpal tissues are enlarged with calcifications with potential pulpitis and pulpal necrosis.9,34,35 Pulp horns may extend to the enamel surface, similar to rickets. 35 The surrounding follicular tissues may demonstrate focal aggregates of basophilic amorphous calcifications called “enameloid conglomerates,” which while not specific for regional odontodysplasia, is highly suggestive of the condition in the correct clinical circumstance (Figures 24 and 25).9,29,34 Normal or hyperplastic odontogenic rests may also be found in the adjacent tissues.
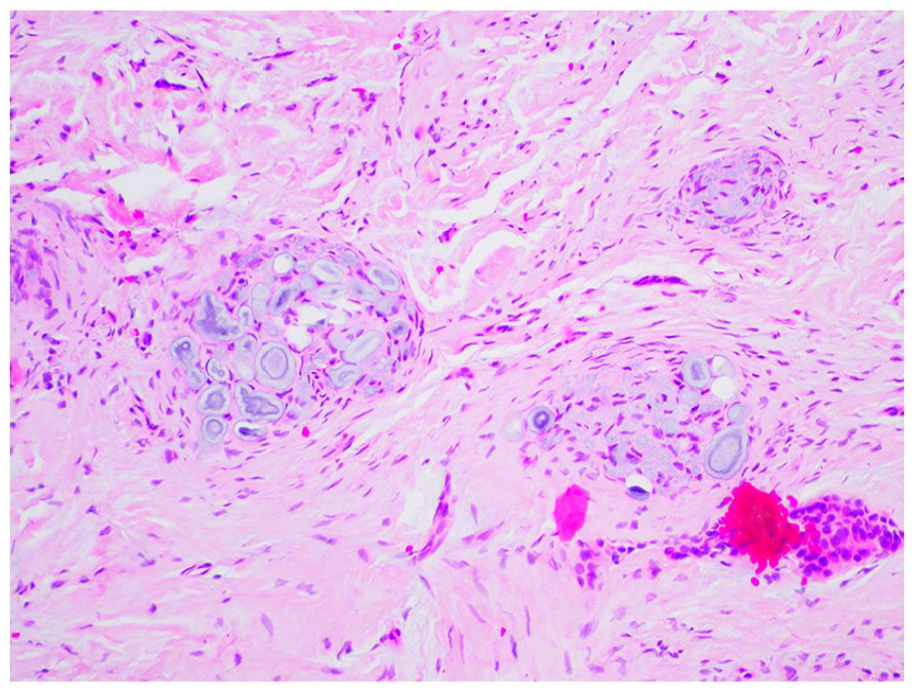
Regional odontodysplasia. Basophilic enameloid conglomerates suspended in fibrous connective tissue.
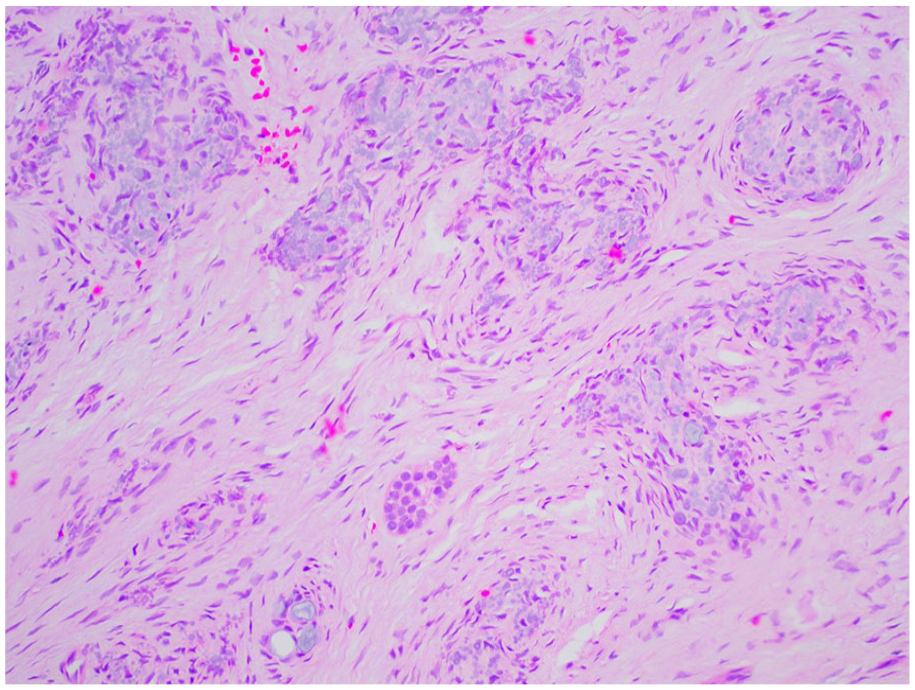
Regional odontodysplasia (ROD). Enameloid conglomerates suspended in follicular tissues with islands of odontogenic epithelium and rests.
Molecular features
None, ROD is not an inherited condition.
Management
Currently, no therapies exist to treat this developmental condition. Maintenance of the existing erupted dentition is accomplished by dental restorative techniques. Unerupted teeth are monitored and may be replaced with dental prostheses. Extraction of hopeless and infected dentition may be indicated.9,34
Conclusion
Histologic examination of dental anomalies in the pediatric patient is typically performed as an adjunct method of diagnosis. Dental features such as color changes, morphologic alterations, and aberrant tooth eruption sequences are valuable findings toward establishing a diagnosis. Radiographic correlation is recommended in assessing potential dental anomalies, starting with dental imaging. Routine decalcification preparations may render histologic examination low yield in the case of suspected enamel defects, such as in amelogenesis imperfecta. A genetic workup may be indicated in patients with a history of a tooth anomaly and additional clinical findings such as musculoskeletal disease, hormone imbalances, and craniofacial deformities.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.