
Abstract
Primary hepatic angiosarcoma (HAS) is an exceedingly rare tumor that is often idiopathic and usually fatal. It typically occurs in a bimodal age distribution, in older adults or very young children. We describe a unique case of HAS arising at an unusual age, in an adolescent who was one of a pair of identical twins affected by cryptogenic cirrhosis and who were found to have germline loss-of-function variant in MAF, a gene that has recently been shown through animal models to be crucial in the specification of liver sinusoidal endothelial cells (LSECs). The case is the first known report of liver disease in the setting of germline MAF variants. It implicates LSEC dysfunction in both non-neoplastic and neoplastic liver disease and lends insight into the importance of LSECs in liver health and homeostasis.
Introduction
Primary hepatic angiosarcoma (HAS) is a rare and aggressive tumor arising from endothelial cells, with a dismal prognosis. It has a bimodal age distribution, with peak incidence in the sixth decade; rare cases occur in very young pediatric patients (average age 3.5, range 1-7), usually in the setting of multifocal infantile hemangiomas. 1 Most cases are idiopathic, although exposure to specific carcinogens like vinyl chloride, arsenic, and Thorotrast 2 are known risk factors and rare association with hemochromatosis has been reported. 3 On occasion, HAS arises in the setting of cirrhosis but their relationship, whether causal or coincidental, has long been a subject of debate.3,4
Here, we present a unique case of hepatic angiosarcoma presenting at an unusual age, arising in one of a pair of adolescent identical twins who were both affected by cryptogenic cirrhosis in the setting of a germline MAF loss-of-function variant and a heterozygous ATP7B variant. C-Maf, encoded by Maf/MAF, has recently been shown in mouse and in vitro in human cells to be critical to the specification of liver sinusoidal endothelial cells (LSECs). 5 We explore how this case, in conjunction with the recent c-Maf studies, sheds light on the relationships between LSECs, non-neoplastic liver disease, and HAS.
Case Report
A 17-year-old Caucasian male with a history of bilateral cataracts and dentinogenesis imperfecta was referred to pediatric gastroenterology with sudden onset abdominal pain and diarrhea. He was otherwise asymptomatic. His family history was notable for having an identical twin brother who also had bilateral cataracts and dentinogenesis imperfecta. Labs were significant for thrombocytopenia, an international normalized ratio of 1.3, as well as a mild elevation of total bilirubin and ammonia. Immunglobin G (IgG) was minimally elevated and smooth muscle antibody (SMA) was low positive (titer 1:80); all other labs were normal. Abdominal computed tomography demonstrated a small nodular liver with evidence of portal hypertension and splenomegaly.
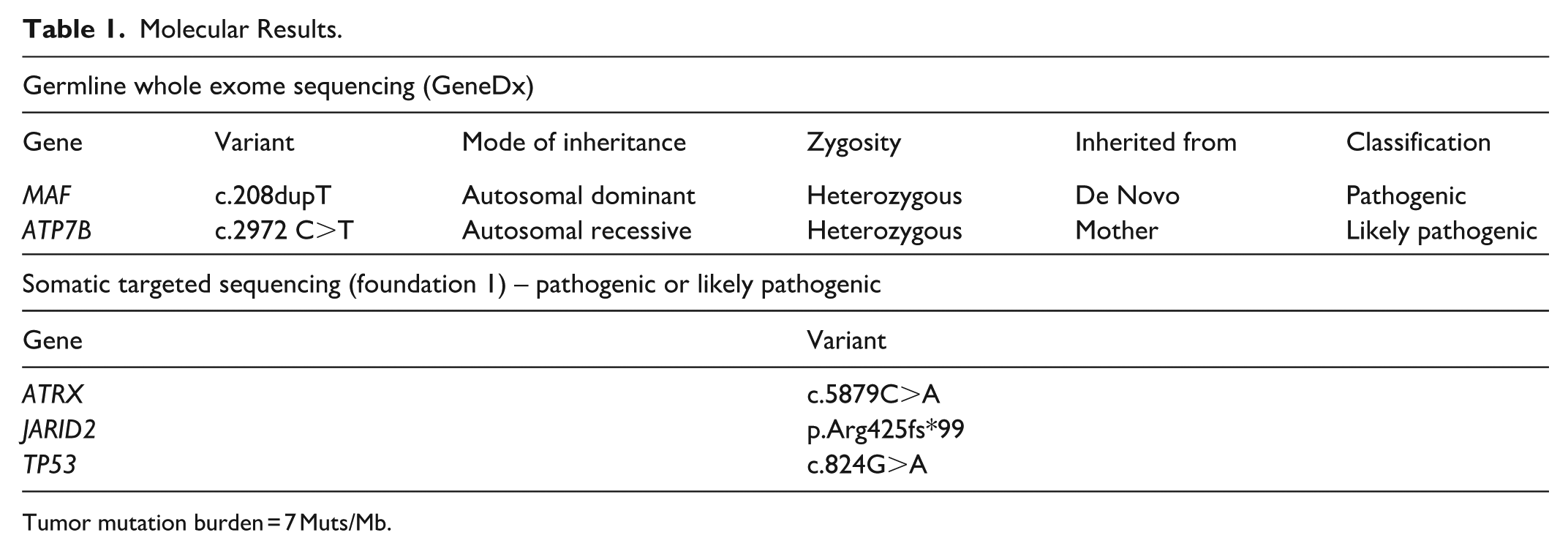
A non-targeted liver biopsy demonstrated hepatic parenchyma without significant inflammation, steatosis, or overt signs of hepatocellular or bile duct injury. There was variable fibrosis with some areas showing no fibrosis and other areas showing advanced fibrosis with nodularity, consistent with cirrhosis (Figure 1(A) and (B)). Some microvascular abnormalities were noted, affecting both fibrotic and non-fibrotic areas. Portal veins, for example, were difficult to visualize in some portal tracts (Figure 1(C)), sinusoids showed partial capillarization, demonstrated on CD34 immunohistochemistry (Figure 1(D)), and alternating hepatic plates were mildly widened and flattened on reticulin stain. There was no ductopenia. Other ancillary stains were unremarkable. The primary diagnosis at that time was cryptogenic cirrhosis. His twin brother who developed similar symptoms, underwent a workup with lab findings similar to his brother’s. His liver biopsy showed similar findings, and he was also diagnosed with cryptogenic cirrhosis. Whole exome sequencing was performed and both siblings were found to have a loss of function de novo mutation of the MAF gene and heterozygosity for a mutation in gene ATP7B (refer to Table 1 for details).
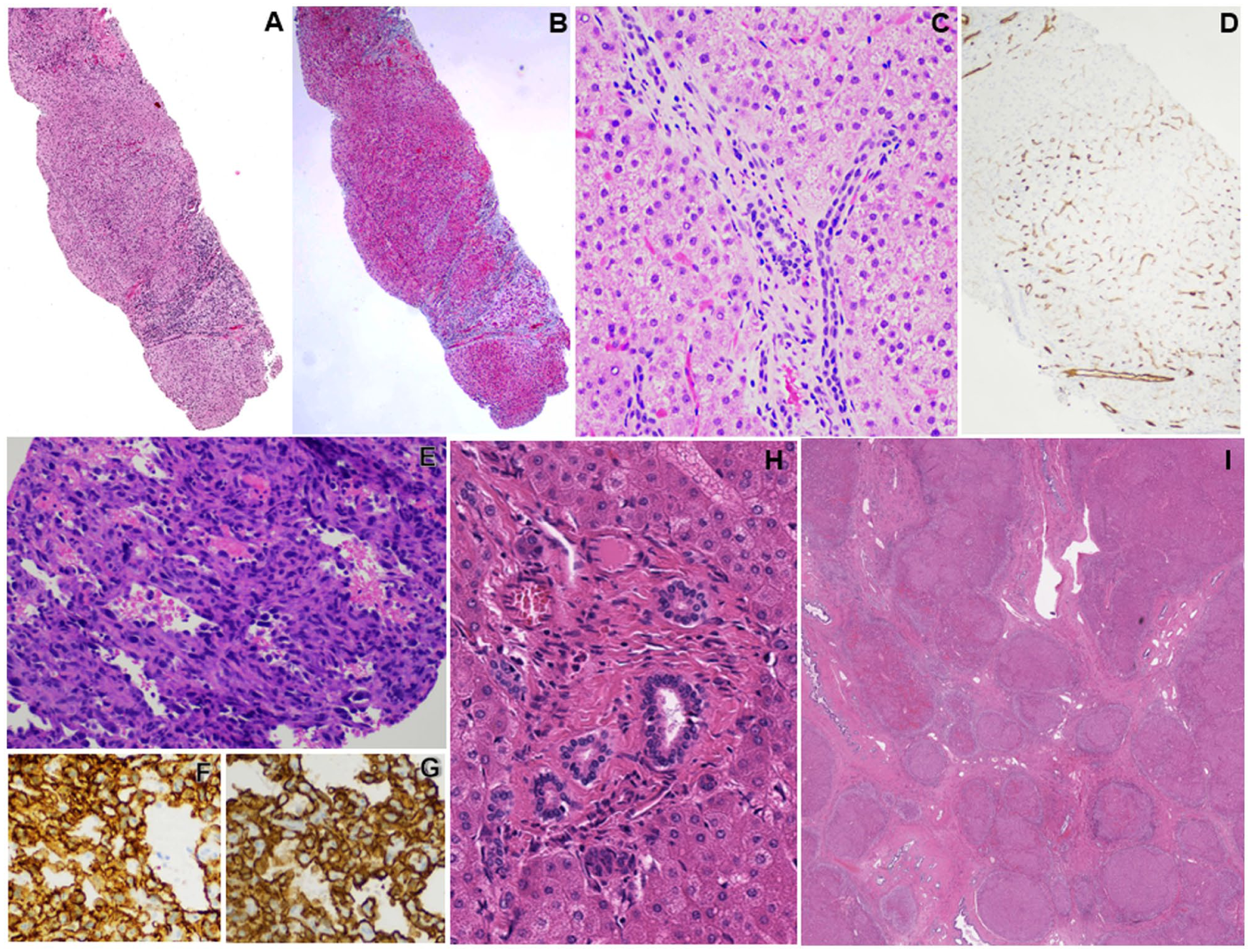
Liver histopathologic findings from both twins. (A-D) Biopsy of the deceased twin’s uninvolved native liver demonstrates areas of nodularity (A, hematoxylin and eosin stained) corresponding to increased fibrosis as demonstrated on Masson trichrome staining (B). Signs of microvascular abnormalities observed include portal tracts missing portal veins (C, hematoxylin and eosin stained) and partial sinusoidal capillarization highlighted by CD34 positivity in Zones 1 and 2 liver sinusoidal endothelial cells (D, immunoperoxidase stain). (E-G) Biopsy of liver tumor from the same twin demonstrating a proliferation of malignant cells with striking pleomorphism and hyperchromasia lining anastomosing vascular spaces (E, hematoxylin and eosin stained), and which are diffusely positive for CD31 and CD34 (F and G respectively, immunoperoxidase stained), consistent with angiosarcoma. (H-I) Explanted native liver from the surviving twin demonstrated similar findings to his twin brother’s native, uninvolved liver, including portal tracts with sclerosis and absent portal vein (H). There were also several areas of nodularity consistent with cirrhosis (I). (H-I, hematoxylin and eosin stained).
Molecular Results.
Tumor mutation burden = 7 Muts/Mb.
A doppler ultrasound of the liver shortly after the biopsy demonstrated 2 areas of focal, heterogeneous nodularity in the left hepatic lobe measuring 1.5 and 4.4 cm, thought to represent regenerative nodules. Five months later, the patient re-presented with ascites. An abdominal MRI demonstrated interval development of multiple masses in the liver (Figure 2).

Magnetic Resonance Imaging of the deceased twin in the coronal (A) and transverse (B) planes show markedly abnormal appearance of the entire liver with numerous infiltrative and heterogeneously enhancing masses, the largest of which was 9.3 cm × 6.4 cm × 7.2 cm.
A biopsy of the liver masses showed a neoplasm composed of epithelioid to spindle cells demonstrating hyperchromasia, marked pleomorphism, and frequent mitoses lining inter-anastomosing vascular-type channels. By immunohistochemistry, the tumoral cells were positive for CD34 and CD31, supporting a diagnosis of angiosarcoma of the liver (Figure 1(E)-(G)). Next generation sequencing on the tumor showed mutations in ATRX, JARID2, and TP53 (Table 1).
Treatment was initiated with paclitaxel, pazopanib, and propranolol. The patient received 4 cycles of chemotherapy with PET scan demonstrating decreased tracer uptake in the liver and no metastases, suggesting partial response of his hepatic tumors. As a result, he was referred for liver transplant evaluation. However, an excisional biopsy from an enlarged omental lymph node during a pre-transplant exploratory laparoscopy confirmed metastatic angiosarcoma. The transplant was cancelled. He was placed on palliative care and died 2 weeks later.
His twin brother did not develop neoplasia and was soon transplanted. The explanted native liver showed findings similar to that seen on both twins’ native liver biopsies, including cirrhosis (Figure 1(H)) and a lack of portal veins in some portal tracts (Figure 1(I)). He is currently alive and doing well, with no evidence of recurrent disease.
Discussion
The occurrence of HAS in adolescence, as in our case, is exceedingly rare – the patient was older than expected for pediatric HAS and did not have a history of infantile hemangioma. He was much younger than the typical adult affected by HAS, suggesting the possibility of a predisposing condition. Indeed, the few cases of HAS reported in the adolescent age group have been with a background of dyskeratosis congenita. 6 The twins’ past medical history of dentinogenesis imperfecta and congenital cataract raises additional suspicion for genetic cause of their chronic liver disease, and potentially one that conferred susceptibility to angiosarcoma.
Of the 2 variants uncovered via the twins’ germline whole exome sequencing, the heterozygous mutation in ATP7B, a gene implicated in Wilson disease, is unlikely to have caused the twins’ cirrhosis given the autosomal recessive inheritance pattern and their lack of clinical or laboratory signs of Wilson disease.
The de novo loss of function MAF variant is compelling, given recent report of the crucial role of Maf in the specification of “liver identity” in liver sinusoidal endothelial cells (LSECs). 5 Further, LSECs are essential to liver function, including the maintenance of hepatic stellate cells in their quiescent stage, preventing fibrosis. 7 The MAF variant is, at the least, responsible for their congenital cataracts, given the importance of MAF in embryonic lens development and that missense MAF germline mutations are a well-established cause of congenital cataract. 8
To our knowledge, liver disease has not yet been reported in association with MAF germline variants. In a recent study, knocking Maf out in endothelial cells in the mouse prevented appropriate liver-specific differentiation of endothelial cells into LSECs. 5 Further, knocking MAF into more generic human ECs in vitro imparted liver-specific characteristics. In the mouse, loss of Maf in ECs and concomitant mis-specified ECs resulted in EC hyperplasia and an exaggerated response of the liver to fibrosis when injured. 5 The relationship between LSEC differentiation and fibrosis have previously been demonstrated in animal models, showing that loss of LSEC specification, manifesting as loss of fenestration, capillarization, and increased vasoconstriction, is an early event that precedes liver fibrosis.9,10 Differentiating LSECs, on the other hand, promotes reversion of hepatic stellate cells to their quiescent state.10,11 It is thus possible that the twins loss of function MAF mutation conferred LSEC dysfunction, resulting in an exaggerated fibrotic response to low level injury that may have resulted from their heterozygous ATP7B mutation.
The microvascular changes in both twins’ native livers are difficult to interpret in the setting of cirrhosis as microvascular changes are often present within significantly fibrotic or cirrhotic livers. Given the heterogeneity of fibrosis in the liver and the presence of microvascular changes even in areas away from advanced fibrosis, however, it remains possible that these changes reflect vascular-related dysfunction due to loss of LSEC specification.
The initiation of angiosarcoma almost certainly followed the development of chronic liver disease and cirrhosis, given that one of the twins developed cirrhosis but not angiosarcoma. Tumorigenesis was likely driven by the pathogenic somatic TP53 and ATRX variants detected in the tumor; ATRX loss is reportedly frequent in primary HAS, and in conjunction with TP53 deficiency confers susceptibility to sarcomas. 12 It is very possible that LSEC dysfunction, as suggested by the mouse model, led to dysregulated growth conferring predisposition to angiosarcoma via somatic mutations.
That EC dysfunction/injury could underlie both chronic liver disease and angiosarcoma has been suggested by other phenomenon, most significantly vinyl chloride related toxicity, which is known to lead to portal hypertension, cirrhosis, and angiosarcoma. 13 In dyskeratosis congenita and X-linked agammaglobulinemia, nodular regenerative hyperplasia, which can result from EC injury, is observed and rare incidences of hepatic angiosarcoma have been reported.6,14 A prior case series of 35 patients with hepatic angiosarcoma included 15 patients with cirrhosis. 4 The rarity of angiosarcoma compared with cirrhosis, as well as their co-presentations in contexts of EC injury suggests that their co-occurrence may be better explained by a common underlying etiology, such as EC dysfunction, injury, or mis-specification. Our current case gives credence to this idea, given the putative function of MAF in LSEC specification.
In conclusion, this case represents the first report of human liver disease associated with an MAF germline variant and, importantly, strongly implicates EC dysfunction/injury as a potential driver of both chronic non-neoplastic liver disease and primary hepatic angiosarcoma, lending mechanistic insight into other known etiologies such as vinyl chloride, dyskeratosis congenita, and hemochromatosis. Given the rapid progression of hepatic angiosarcoma and vital importance of timely intervention, vigilance is necessary especially in cases that may involve EC dysfunction, including cryptogenic chronic liver disease. More broadly, the case adds to increasing evidence that LSECs are “primary” cells of the liver, essential to its health and function.
Footnotes
Ethics Considerations
Written informed consent was obtained for the publication of this case report.
Author Contributions
M.P.D., R.W., A.B., and S.Y.T. contributed to study concept and design, analysis and interpretation of data, and drafting of the manuscript; G.G. contributed acquisition, analysis, and interpretation of data; V.G. contributed acquisition of data; all authors critically reviewed and revised the manuscript for important intellectual content.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.