
Abstract
Background:
Erythropoietic Protoporphyria (EPP) is a rare inherited disorder of heme biosynthesis caused by pathogenic variants in FECH. Although most patients present with cutaneous photosensitivity in childhood, clinically significant liver disease is uncommon, and severe fibrosis in early childhood is rarely reported.
Case:
A 2-year-old girl presented with recurrent photosensitivity and erythematous swelling after sun exposure. Laboratory evaluation revealed markedly elevated erythrocyte protoporphyrin (2406 µg/dL) and elevated plasma protoporphyrin. Genetic testing identified compound heterozygous FECH variants: c.901_902del (p.Trp301Alafs*23) and c.801G>A (p.Met267Ile). Liver enzymes were significantly elevated. Following liver biopsy demonstrated stage 3 bridging fibrosis with crystalline deposits in hepatocytes consistent with protoporphyric hepatopathy. The patient subsequently underwent hematopoietic stem cell transplantation with improvement of protoporphyrin levels and liver enzymes. However, the post-transplant course was complicated by severe infections, acute respiratory distress syndrome, and multiorgan failure, resulting in death.
Conclusion:
This case highlights rapidly progressive liver fibrosis in a toddler with EPP and emphasizes the importance of early hepatic monitoring in pediatric patients with markedly elevated protoporphyrin levels and rare FECH variants.
Introduction
Erythropoietic protoporphyria (EPP) is a rare inherited disorder of heme biosynthesis most commonly presenting with painful, non-blistering cutaneous photosensitivity following sun exposure. 1 It is caused by pathogenic variants in the FECH gene, which encodes ferrochelatase, the enzyme catalyzing the insertion of iron into protoporphyrin. The pathogenic variants reduce ferrochelatase activity, which in turn causes accumulation of protoporphyrin IX in erythrocytes and plasma. Subsequently, the excess deposition of protoporphyrin IX in the skin and liver underlies the characteristic photosensitivity and may also contribute to anemia and progressive hepatic dysfunction.2,3
The liver involvement by accumulation of protoporphyrin IX is referred to as protoporphyric hepatopathy, which starts with cholestatic liver injury and may progress to severe liver failure. 4 The pathophysiology of liver damage in EPP involves the hepatotoxic accumulation of protoporphyrin within hepatocytes and bile canaliculi, triggering oxidative injury and bile stasis. Histologically, the presence of birefringent Maltese cross-shaped protoporphyrin crystals under polarized light microscopy is a hallmark of this process. 5,6
While liver involvement has been documented in EPP, significant hepatic disease—including cholestasis, fibrosis, and liver failure—typically develops later in life and affects only a minority of patients. 7 Early onset of liver fibrosis during toddlerhood is even rarer. Here, we report a case of a 2-year-old female with EPP who developed stage 3 liver fibrosis 2 months after diagnosis of EPP to underscore the importance of proactive hepatic monitoring and review the previous reported pediatric EPP with liver fibrosis.
Case Presentation
A 2-year-old female was initially presented with recurrent episodes of facial and extremity swelling, along with erythematous skin lesions triggered by sunlight exposure since May 2023. The genetic testing (May 2025) following the most recent symptom onset revealed compound heterozygosity for a pathogenic frameshift variant, c.901_902del (p. Trp301Alafs*23), with a likely pathogenic missense variant, c.801G>A (p. Met267Ile) in the FECH gene.
At the time of diagnosis, laboratory tests showed significantly elevated levels of metal-free erythrocyte protoporphyrin (2406 µg/dL; normal <20), plasma protoporphyrin (40.5 µg/dL; normal <1), and zinc erythrocyte protoporphyrin within the reference range (48 µg/dL; normal <60). Her liver function tests (LFTs) showed significantly increased AST (306 U/L, normal 9-32U/L) and ALT (273 U/L, normal 7-33 U/L), suggesting the hepatopathy related to EPP. Her total bilirubin was normal (0.7 mg/dL, normal 0.2-1.2 mg/dL), direct bilirubin was mildly increased (0.4 mg/dL, normal 0.1-0.2 mg/dL) and alkaline phosphatase was within the reference range (277 U/L, normal 156-369 U/L).
Initial management included cholestyramine, ursodeoxycholic acid (initiated in May 2025). The patient initially demonstrated an improvement in transaminase levels. However, rapid progress occurred 1 month later (June 2025) with rising AST (153 U/L, normal 9-32U/L) and ALT (172 U/L, normal 7-33 U/L), concerning progressive hepatopathy. The patient was arranged for prompted imaging-guided liver biopsy for histological staging (July 2025). The biopsy revealed stage 3 bridging fibrosis (portal-portal) with mainly hepatocellular deposition of crystalline material (Batts-Ludwig system), scattered in the lobules and portal tracts. Under polarized light, the crystalline material demonstrated cross-shaped birefringence (Maltese cross-shaped crystals), which is characteristic of protoporphyrin accumulation. The portal tracts demonstrated non-specific lymphocytic infiltrates, with no significant bile duct damage or bile duct proliferation. No significant lobular inflammation and cholestasis were identified. Other potential etiologies, like viral hepatitis, drug-induced liver injury, autoimmune hepatitis, sclerosing cholangitis, and other causes of intrahepatic cholestasis, were excluded through appropriate testing. Together with the histological features, these results argue against concurrent diseases and support EPP involvement as the main etiology of hepatic injury (Figure 1(A)-(D)).
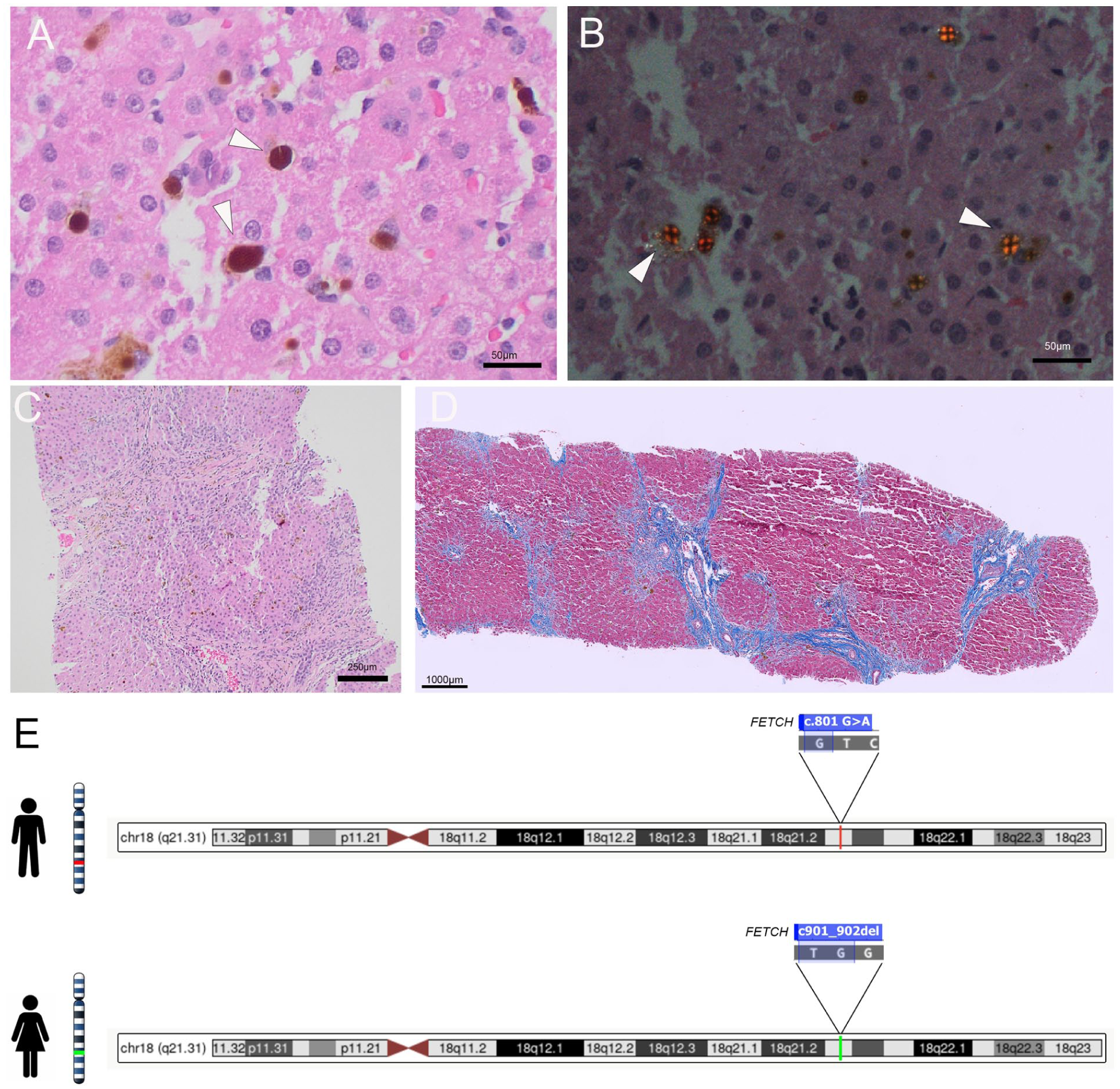
Liver biopsy and genetic test. (A) H&E demonstrated brown crystalline deposition (arrowheads). (B) Maltese cross-shaped crystals under polarized light, confirming the presence of protoporphyrin (arrowheads). (C) H&E suggested stage 3 bridging fibrosis. (D) Masson Trichome stains highlighted the bridging fibrosis (blue). (E) Targeted familial variants testing in the patient’s parents suggested that the 2 variants were inherited from each parent, respectively.
Despite advanced-stage fibrosis, the patient still maintained preserved hepatic synthetic function, with normal ammonia, INR, and bilirubin level, and no clinical evidence of ascites or encephalopathy. Liver transplant was not considered before the bone marrow transplant.
The patient then received an urgent bone marrow transplant. Her plasma protoporphyrin levels were trending down from 58.4 µg/dL to 1.1 µg/dL (normal <1), suggesting her EPP was significantly corrected. Both her AST and ALT decreased to the normal range within 1 week after bone marrow transplantation, indicating improvement of hepatopathy. One week before transplantation, total bilirubin and direct bilirubin increased to 4.0 mg/dL and 2.8 mg/dL, respectively; however, both levels decreased within 1 week post transplantation. Her alkaline phosphatase remained within normal range throughout the peri-transplantation period. The post-transplant course was complicated by bacteremia, cytomegalovirus (CMV) viremia, acute respiratory distress syndrome (ARDS), and acute kidney injury. Despite aggressive supportive treatment, the patient died due to multiorgan failure.
Discussion
EPP is an autosomal recessive inherited disease, genetically characterized by a loss-of-function FECH mutation in trans with a common hypomorphic FECH genetic variant (over 90% with FECH c.315-48T>C). This combination leads to a significantly decreased activity of ferrochelatase (<35% enzymatic activity), and subsequent accumulation of protoporphyrin IX causes a series of symptoms. 8 Homozygosity for FECH c.315-48T>C does not cause EPP, as this common low-expression FECH allele is present in ~10% of the Caucasian population, and biallelic c.315-48T>C still preserves enough ferrochelatase activity. 3 In our patient, the c.901_902del (p. Trp301Alafs*23) variant creates a premature translational stop signal in the FECH gene. It is expected to result in an absent or disrupted protein. This loss-of-function variant has been observed in individuals with EPP before.2,9,10 Interestingly, the other allele our patient harbors (c.801G>A) was classified as a variant of uncertain significance, however, this has been reported in multiple cases of EPP in individuals who have 2 rare pathogenic variants.3,11,12 A prior in vitro study showed that heterodimers composed of the wild-type FECH and c.801G>A exhibited reduced FECH stability and less than 20% of activity, and homodimers of c.801G>A totally abolished the enzymic activity, 12 suggesting this rare allele might be more hypomorphic than c.315-48T>C. Balwani et al 13 described a significant correlation between erythrocyte protoporphyrin levels and earlier age at onset, decreased sun tolerance, and increased risk of liver dysfunction. This hypomorphic variant might account for her extremely high erythrocyte protoporphyrin, which would increase her risk for EPP-related liver disease. Further targeted familial variant testing of the patient’s parents suggested that the c.901_902del (p. Trp301Alafs*23) variant was inherited from the father, and the c.801G>A (p. Met267Ile) variant was inherited from the mother, confirming that the variants are in trans (Figure 1(E)).
One of the differential diagnoses of EPP is X-linked dominant protoporphyria (XLDPP). Clinically distinguishing between the 2 can be challenging as they can have similar traits. Genetically, XLDPP is caused by genetic changes in ALAS2, which can be detected by a genetic test. However, the metal-free and zinc-bound protoporphyrin ratio within erythrocytes can also help differentiate. An elevated total erythrocyte protoporphyrin level that is predominantly metal-free is diagnostic of EPP, whereas XLDPP usually has a higher ratio of zinc-bound protoporphyrin to metal-free protoporphyrin. Despite the different etiology, the liver histopathology in both diseases is the same.
Other key differentials include progressive familial intrahepatic cholestasis (PFIC), biliary atresia, and drug-induced cholestasis, each with distinct—though sometimes overlapping—histopathologic features. In PFIC, especially PFIC1 and PFIC2, the early disease phase demonstrates intralobular cholestasis with minimal ductular reaction; with progression, hepatocellular ballooning, giant cell transformation, and periportal/pericellular fibrosis can be present. In contrast, PFIC3 more commonly shows ductular reaction, portal fibrosis, and bile duct injury. 14 In biliary atresia, the hallmark findings include marked, florid ductular proliferation (commonly accompanied by bile plugs), portal edema and fibrosis (often progressing to bridging fibrosis); bile duct injury and eventual duct loss may be seen as the disease advances. 15 Drug-induced cholestasis is more heterogeneous but typically shows canalicular and hepatocellular cholestasis with variable lobular disarray, hepatocellular ballooning, and mixed inflammatory infiltrates, often with eosinophils, suggesting a hypersensitivity pattern. 16 Recognizing the degree of ductular reaction, portal fibrosis, and inflammatory pattern, together with clinical context, is critical for distinguishing these entities on histologic grounds.
In EPP, liver failure can progress rapidly. As protoporphyrin is insoluble and lipophilic and is excreted in the bile rather than urine, the accumulation of protoporphyrin IX precipitates in hepatocytes and bile canaliculi causes the downstream oxidative stress and inflammatory cascade resulting in hepatocyte apoptosis and necrosis. The injured liver would further increase the protoporphyrin precipitates, and this vicious cycle contributes to the rapid progress of liver failure. Our patient’s liver ultrasound in May 2025 was normal. In June 2025, however, the ultrasound showed a moderate risk of clinically significant fibrosis, which was confirmed by histology through subsequent biopsy despite continuous treatment with cholestyramine and ursodiol. Once it progresses to hepatic failure, an orthotopic liver transplant is inevitable.
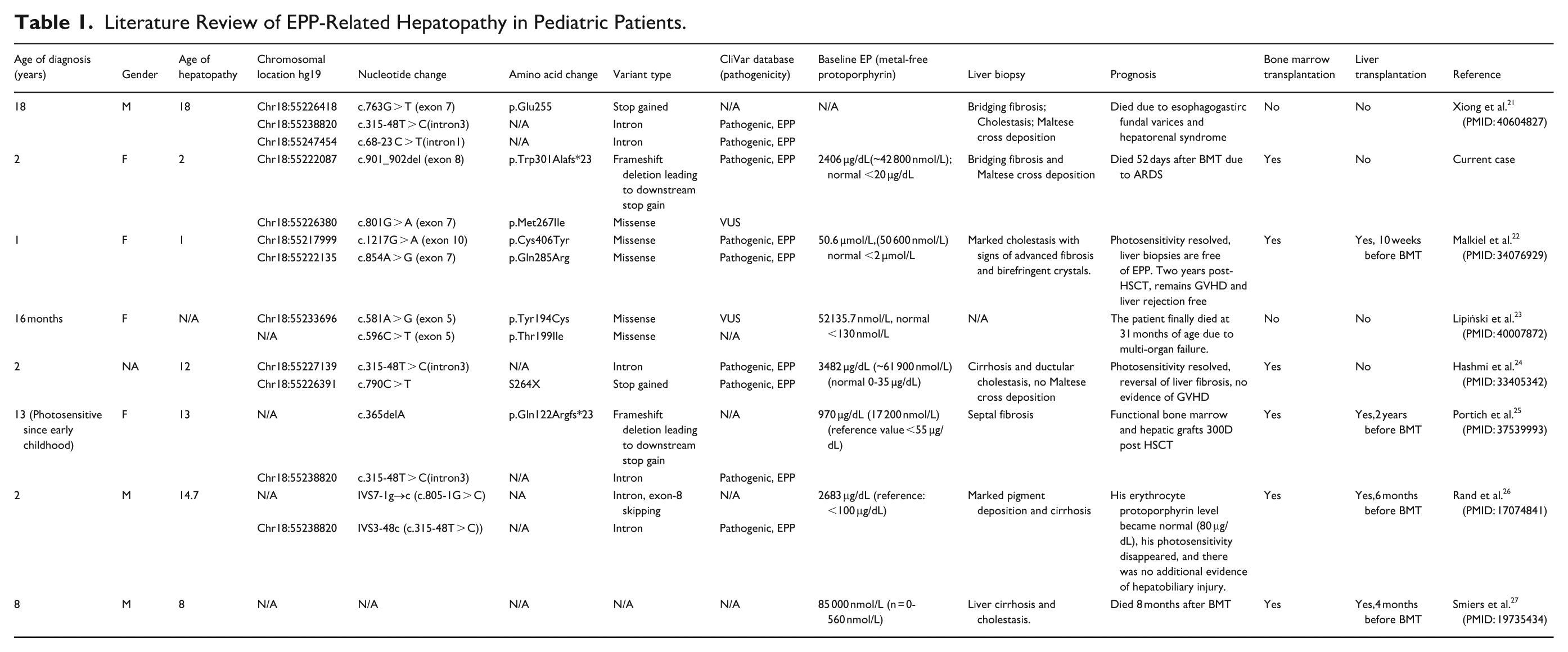
Regardless of the severe liver damage caused by EPP as the disease progresses, protoporphyric hepatopathy is rarely recognized at an early stage, and the mechanism is still unknown. Liver fibrosis is even more uncommonly seen in early toddlerhood. To summarize the characteristics of this rare clinical presentation, we reviewed the English literature indexed in PubMed and Scopus using the following search terms: “erythropoietic protoporphyria,” “EPP,” “liver fibrosis,” “liver disease,” “pediatric,” and “children.” Eight pediatric EPP patients with protoporphyric hepatopathy were identified (including the case reported here, Table 1). Seven cases developed liver fibrosis confirmed by histology (one case presented with progression of hepatosplenomegaly with rising parameters of cholestasis but no liver biopsy performed), and 2 of the 7 cases progressed to fibrosis at toddlerhood (mean age: 21 months; range, 1-1.5 years). Their mean metal-free/erythrocyte protoporphyrin value was 2628 µg/dL. The association between higher erythrocyte protoporphyrin values (>1517 μg/dL) and cirrhosis has been established among EPP patients.17,18 Therefore, the exceedingly elevated erythrocyte protoporphyrin in these pediatric patients might put them at high risk of progressing to early liver fibrosis. The mechanism for such a high level of erythrocyte protoporphyrin is still unknown, but it is hypothetically associated with the rare FECH variants. More studies focusing on the enzymic activity with FECH variants are needed to illuminate the mechanism.
Literature Review of EPP-Related Hepatopathy in Pediatric Patients.
Even though orthotopic liver transplantation is effective for treating liver failure, subsequent hematopoietic stem cell transplantation is inevitable, as it can correct the underlying protoporphyrin overproduction in the native bone marrow. Both cases reported in toddlers aged 2 years or under underwent bone marrow transplantation; one had previously undergone liver transplantation and had a favorable outcome, whereas the other died of ARDS following bone marrow transplantation, indicating the high risk and challenge of bone marrow transplantation in patients with early liver fibrosis.
Besides the invasive treatment, studies have shown that bitopertin, an orally bioavailable small-molecule inhibitor of glycine transporter 1 (GLYT1), conducts a substantial reduction in RBC protoporphyrin IX accumulation in the EPP mouse model. 19 Furthermore, 3 case studies demonstrated that the treatment of cimetidine was effective to reduce photosensitive in EPP pediatric patients. 20 These studies are promising, but more research on pharmaceutical treatment is still needed.
Conclusion
EPP with advanced liver disease in early childhood is rarely reported. Our case demonstrated rapidly progressive EPP-related hepatopathy at the age of 2 years. The high level of erythrocyte protoporphyrin and the rare pathogenic FECH variant in this case likely account for the early-onset severe hepatopathy. Although bone marrow transplant is effective in correcting the protoporphyrin IX (PPIX) accumulation and stabilizing hepatopathy, the procedure carries high risks of mortality. Noninvasive therapies are currently under investigation and may represent alternative treatment options for patients with risk factors.
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.