
Abstract
We report an unusual case of juvenile myelomonocytic leukemia (JMML) in a 4-year-old male who had persistent monocytosis and intermittent anemia and thrombocytopenia beginning at 1 year of age. Given a history of recurrent skin infections, the patient initially received an evaluation for a primary immune disorder, but workup was inconclusive. Bone marrow examination was performed to further evaluate persistent and progressive cytopenias. The findings fulfilled the criteria for JMML with peripheral blood monocytosis ≥ 1×109/L, blast/promonocyte percentage of <20% in peripheral blood and bone marrow, clinical evidence of organ infiltration with hepatosplenomegaly, and absent BCR::ABL1 fusion and KMT2A gene rearrangements. Additionally, next generation sequencing (NGS) studies performed on the bone marrow aspirate revealed multiple acquired somatic mutations, including a KRAS G12D mutation, meeting criteria for the diagnosis of JMML in the context of this case. A novel ETV6 R369W mutation in the setting of JMML was also detected, which may explain the striking megakaryocytic dysplasia in the bone marrow beyond what is typically seen in JMML. This case showed interesting features overlapping with Ras-associated autoimmune leukoproliferative disorder (RALD), including monocytosis and recurrent infections. Furthermore, this case demonstrated a need for more definitive criteria for distinguishing JMML from RALD.
Keywords
Introduction
JMML is a rare myeloproliferative neoplasm of early childhood with peripheral granulocytosis and monocytosis. 1 This disorder was previously classified under the WHO classification as a myelodysplastic syndrome/myeloproliferative neoplasm overlap. 2 The various disorders meeting the current JMML criteria are now understood to be characterized by RAS-activating genetic lesions in the hematopoietic stem cell. These lesions are acquired during the perinatal period or are inherited in the context of a developmental disorder associated with RAS pathway constitutional activation (RASopathy), including Neurofibromatosis type 1 (NF1), Noonan syndrome, and CBL-syndrome (CBL). Canonical RAS pathway mutations (KRAS, NRAS, PTPN11, NF1, CBL) are somatically acquired in 55% to 65% of cases. Secondary mutations are most commonly detected in SETBP1, ASXL1, EZH2, or SH2B3 or in additional RAS pathway genes. RAS pathway mutations produce increased intracellular signaling in hematopoietic cells leading to uncontrolled, unchecked proliferation of granulocytes and monocytes in a manner that is typical of myeloproliferative neoplasms. In contrast, the pathogenesis in myelodysplastic syndrome involves somatically acquired chromosomal abnormalities and mutations in various genes involved in DNA replication, leading to ineffective hematopoiesis and cytopenias. In children, childhood myelodysplastic neoplasm (myelodysplastic syndrome) may arise in patients with germline predisposition syndromes characterized by genomic instability or germline mutations that increase the propensity for myeloid malignancy. 1
RAS-associated autoimmune leukoproliferative disorder (RALD) is a rare intrinsic lymphocyte apoptosis defect associated with somatic gain-of-function mutations in KRAS or NRAS. It is a nonmalignant clinical syndrome initially identified in a subset of patients with presumptive autoimmune lymphoproliferative syndrome (ALPS). Patients present with persistent monocytosis that is often associated with leukocytosis, lymphoproliferation, and autoimmune phenomena. The course is typically indolent, sometimes with spontaneous resolution.3,4 JMML with somatically mutated NRAS or KRAS shares overlapping features with RALD: leukocytosis with immature granulocytes, persistent relative/absolute monocytosis, hypergammaglobulinemia and autoimmune antibodies, lymphadenopathy and splenomegaly, skin and gastrointestinal manifestations, increased hemoglobin F for age, increased serum vitamin B12 levels, hypersensitivity to GM-CSF, and normal karyotype (JMML 65%, RALD 100%).3,4 Rare cases of RALD have been reported to progress to hematopoietic malignancy, either JMML or acute myeloid leukemia (AML). A lack of definitive criteria for distinguishing RALD from JMML in normal karyotype cases may pose a diagnostic dilemma.3 -5
Case Report
The patient is a 4-year-old male who presented to our institution for pre-transplant evaluation due to clinical concern for childhood myelodysplastic neoplasm (myelodysplastic syndrome), based upon findings from a bone marrow biopsy performed 3 months prior. The clinical history was notable for recurrent Staphylococcal skin abscesses. There was a verbal report of a family history of immune deficiency, but the specific diagnoses or other details were not available. At this presentation, the CBC/differential showed leukopenia (WBC 3900/uL) with neutropenia (300/uL) and monocytosis (2000/uL), normochromic, normocytic anemia (hgb 10.0 g/dL), and thrombocytopenia (61 × 103/uL). Further review of the patient’s chart revealed monocytosis and intermittent anemia and thrombocytopenia dating to 1 year of age. An anemia workup demonstrated increased hemoglobin F (3.1%) and an increased serum B12 greater than 1500 pg/mL (180-914 pg/mL). A negative direct antiglobulin test (DAT) showed no evidence of autoimmune hemolysis. A previous immune deficiency evaluation had shown increased serum IgG 1430 mg/dL (540-1130 mg/dL). A lymphocyte subset panel illustrated increased absolute CD8+ T cells 2919 cells/uL (200-1800 cells/uL) and a decreased CD4:CD8 ratio 0.83 (0.9-2.9). Other laboratory studies for bone marrow failure and primary immune deficiency were unrevealing. Imaging studies showed mild hepatosplenomegaly but no lymphadenopathy.
A bone marrow examination was performed for pre-transplant evaluation. The bone marrow core biopsy was hypercellular for age (>95%). The bone marrow aspirate smear morphology demonstrated an increased myeloid:erythroid ratio of 7.3 and increased, atypical monocytes (30.5%) with 11.0% monoblasts/promonocytes. Immunohistochemical staining for CD34 performed on the core biopsy showed a similar blast percentage with 8.5% CD34-positive blasts. Megakaryocytes were increased in number with small, hypolobated, and dysplastic megakaryocytes, including cells with widely spaced nuclei, seen on both the aspirate smears and the core biopsy (Figure 1). The concurrent peripheral blood smear showed 52.8% atypical monocytes similar in morphology to the bone marrow aspirate smear morphology. No circulating blasts or promonocytes were identified (Figure 2). The prior bone marrow examination performed 3 months earlier, which was reviewed concurrently, showed similar features to the in-house bone marrow, including atypical monocytosis (26.5%), 5% monoblasts/promonocytes, and hypercellularity (100%).
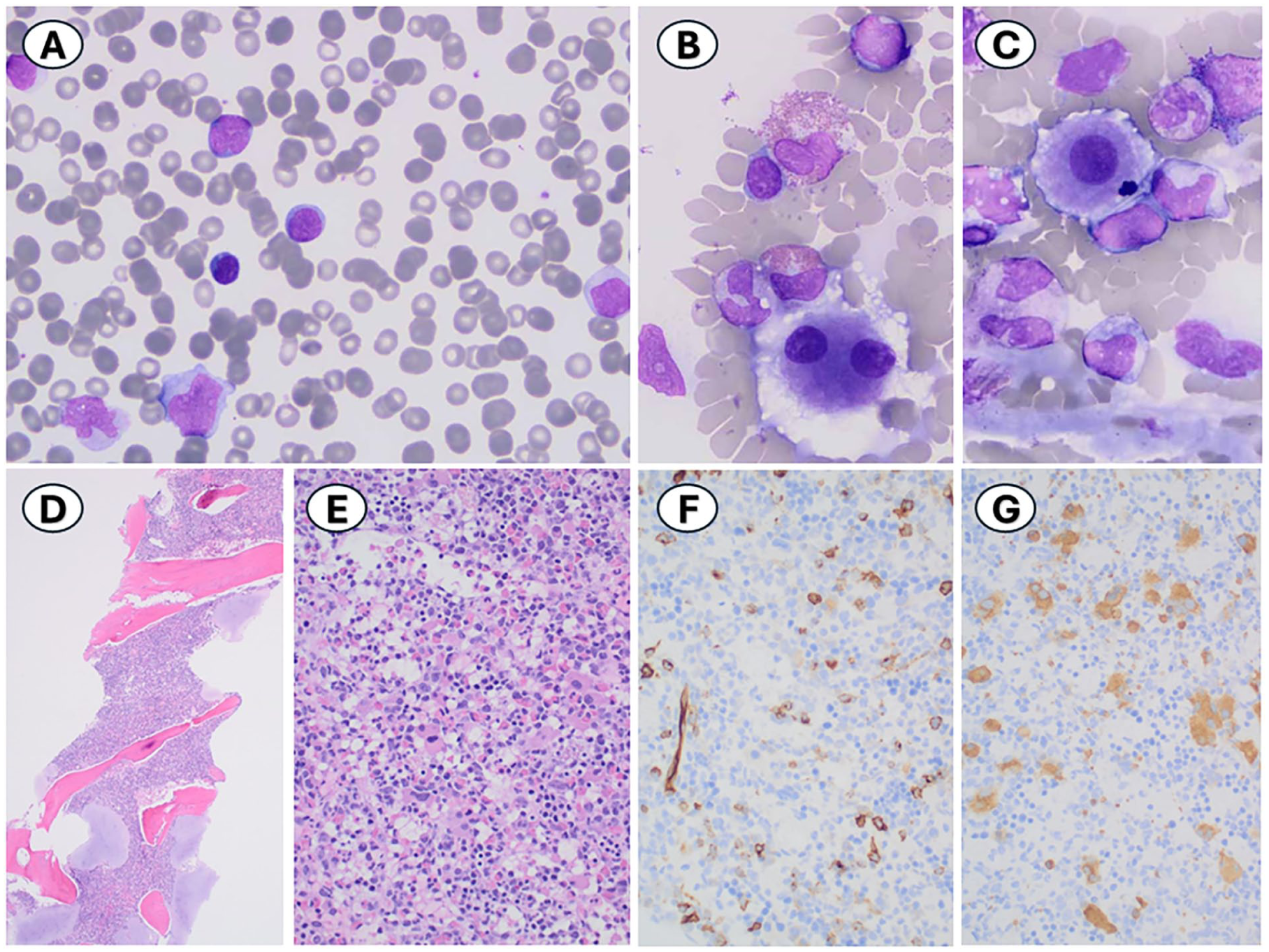
Bone marrow aspirate smears showing (A) atypical monocytes (30.5%) and monoblasts/promonocytes (11.0%), Giemsa stain, 1000×, and (B and C) small, hypolobated megakaryocytes, including cells with widely spaced nuclei, Giemsa stain, 100×. Bone marrow core biopsy showing (D) hypercellular bone marrow (>95%), H&E stain, 40×; (E) increased myeloid:erythroid ratio, H&E stain, 100×; (F) increased CD34-positive blasts (8.5%) on CD34 immunohistochemical stain, 400×; and (G) increased, dysplastic megakaryocytes on CD61 immunohistochemical stain, 400×.
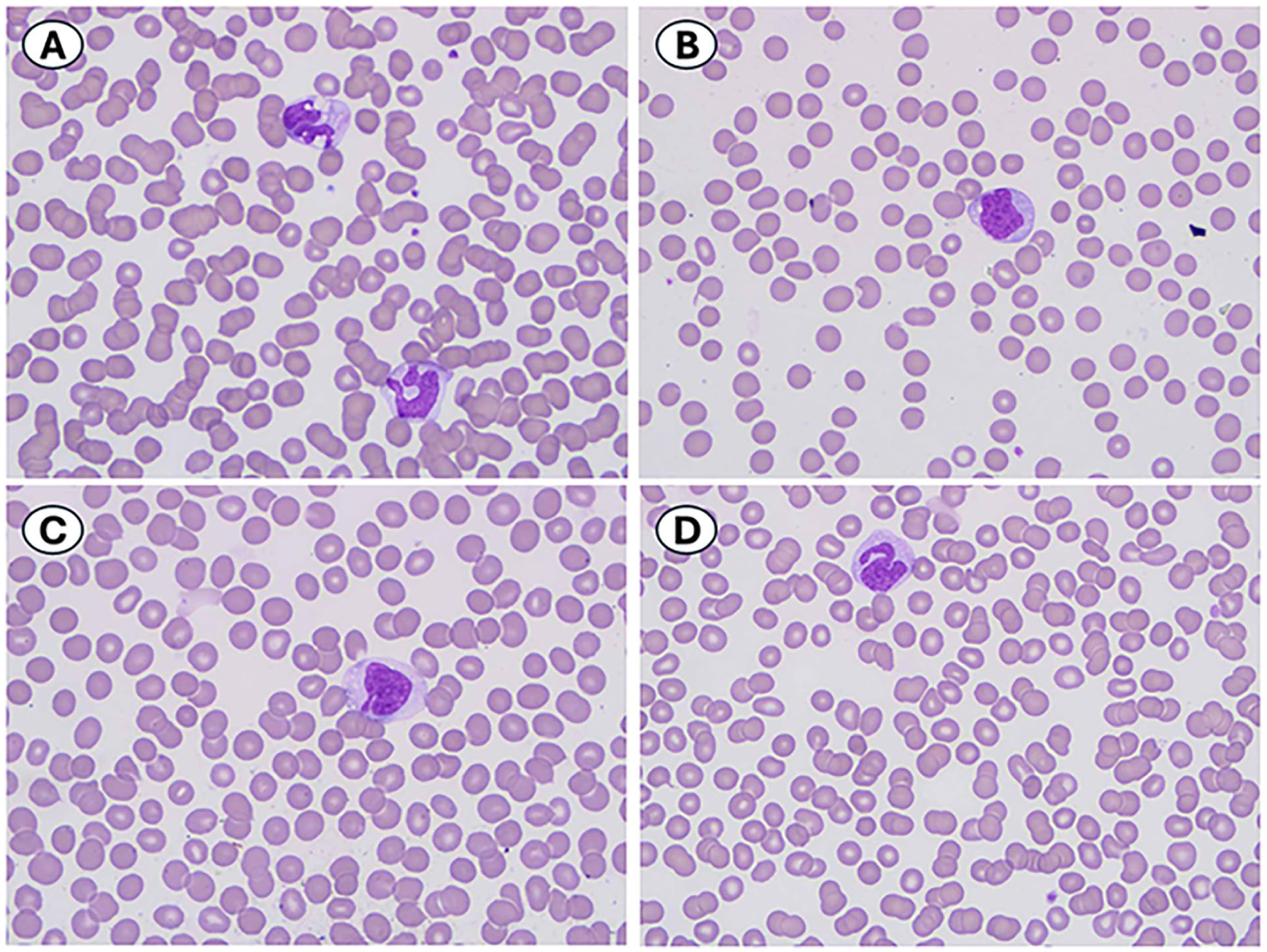
(A-D) Peripheral blood smear showing atypical monocytes (52.8% of leukocytes), Giemsa stain, 1000×.
Flow cytometry studies performed on bone marrow aspirate showed 5% CD45(dim)-positive blasts with heterogeneous CD34 expression and aberrant, heterogeneous CD56 and CD7 expression, as well as monocytes and granulocytes with aberrant CD56 expression. There was a prominent population of CD4- CD8- T cells (16% of CD3+ T cells) which were not further evaluated. B cells were not increased in number, and mature B cells were polyclonal, without aberrant marker expression (Figure 3).
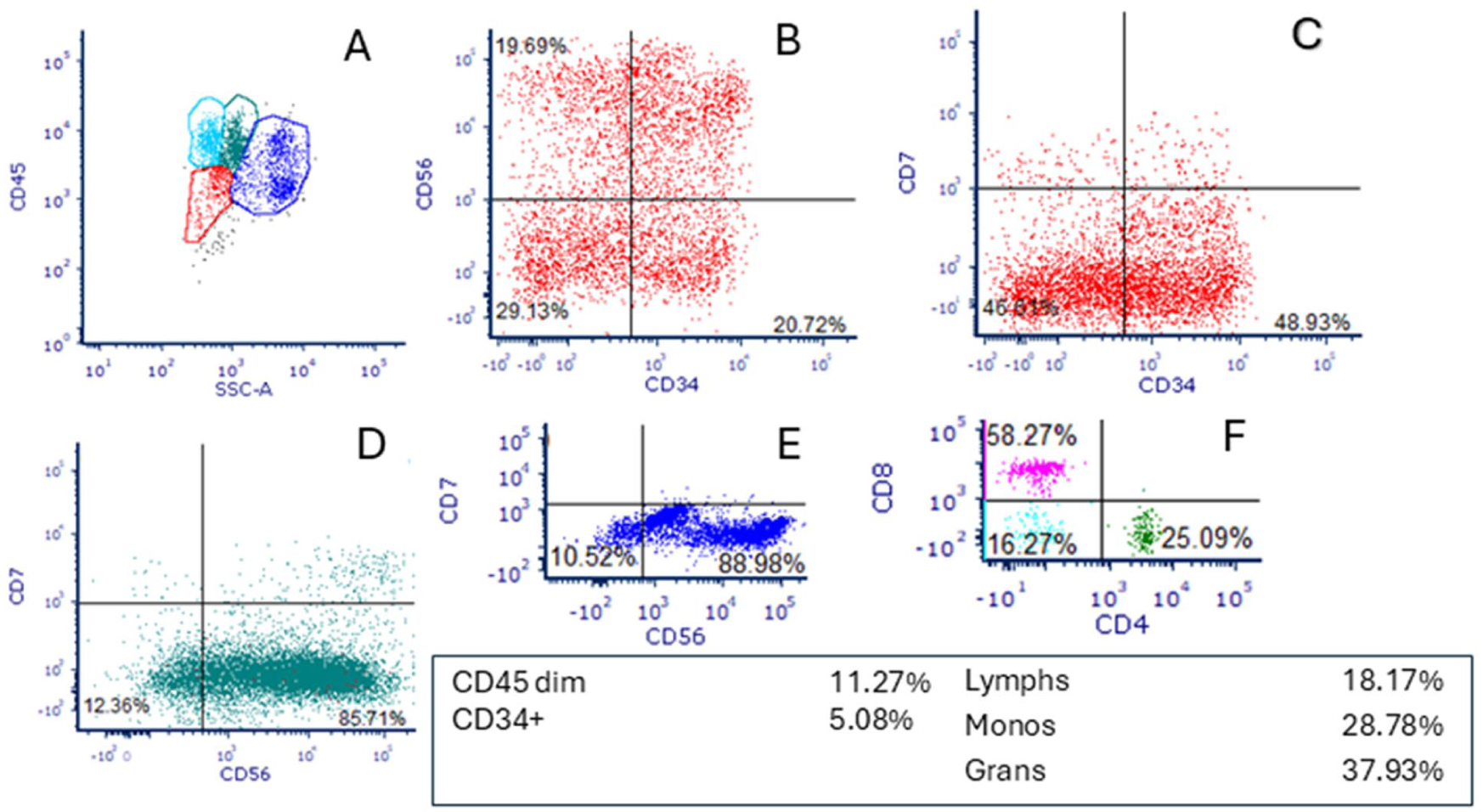
Flow cytometry immunophenotyping studies of bone marrow aspirate showing (A) CD45 versus side scatter histogram with increased CD45 dim+ cells (red) and increased monocytes (green); (B and C) blasts with heterogeneous CD34 expression and aberrant, heterogeneous CD56 and CD7 expression (5% of events); (D) monocytes and (E) granulocytes with aberrant CD56 expression; and (F) increased CD4- CD8- T cells (16% of CD3+ T cells).
Cytogenetic studies performed on bone marrow aspirate demonstrated monosomy 7 (45,XY,-7[20]). FISH studies for 11q23 (KMT2A) were negative for rearrangement. A next generation sequencing panel performed on bone marrow aspirate revealed pathogenic, somatic mutations in ASXL1, KRAS, and ETV6 and a mutation of uncertain significance in JAK3 (Table 1). A targeted germline genetic testing panel was performed to evaluate for mutations predisposing to myeloid neoplasms on cultured buccal mucosa. This panel did not identify any pathogenic variants, including the ETV6 and KRAS variants identified in the bone marrow, confirming that these are somatic variants. The panel was also negative for pathogenic mutations in GATA2, SAMD9, and SAM9L, which may be seen in germline predisposition syndromes associated with monosomy 7. 1
Molecular Mutations Detected in NGS Sequencing Studies.

The diagnosis rendered was juvenile myelomonocytic leukemia (JMML) with 11% blasts/promonocytes. The case met criteria for JMML 1 with the following criteria: peripheral blood monocytosis ≥1 × 109/L, blast/promonocyte percentage of <20% in peripheral blood and bone marrow, clinical evidence of organ infiltration with hepatosplenomegaly, the absence of BCR::ABL1 fusion and KMT2A gene rearrangements, and the presence of a clonal somatic KRAS G12D mutation.
The patient underwent allogenic stem cell transplant and was in remission 2 years post transplant.
Discussion
We report a case of JMML with striking megakaryocyte dysplasia, which contributed to the original concern for childhood myelodysplastic neoplasm (myelodysplastic syndrome) on an outside bone marrow biopsy performed 3 months prior. The megakaryocytes were observed to be markedly increased in number in addition to being dysplastic, whereas megakaryocytes are more commonly decreased in JMML. 1 Nonetheless, the overall constellation of findings, including monocytosis, bone marrow hypercellularity, hepatosplenomegaly, and a clonal somatic KRAS mutation, was more supportive of JMML than myelodysplastic syndrome.
Some myeloid neoplasms are unique to the pediatric age group or have unique aspects that differ from their adult counterparts and require special consideration during diagnostic workup. A number of confounding factors may render a diagnosis more challenging and less straightforward in pediatric patients. One confounding factor is the existence of syndromes having germline mutations that may also be seen as somatically acquired mutations in various myeloid malignancies. In our case, a novel somatic ETV6 R369W mutation in the setting of JMML was detected. The ETV6 gene, located on the short arm of chromosome 12, encodes a transcription factor involved in growth and differentiation of hematopoietic cells, particularly megakaryocytes.6 -11 Mutations in ETV6 have been described in association with various hematologic malignancies.6 -13 Germline ETV6 R369W mutations have been noted in rare cases of familial ETV6-related thrombocytopenia (ETV6-RT).9,13 Germline mutations of ETV6 have generally been reported to be associated with megakaryocyte hyperplasia and small, hypolobated megakaryocytes/dysmegakaryopoiesis in the bone marrow, in the absence of malignancy.7 -10 Interestingly, there are also rare reports of CMML, the adult chronic myeloid malignancy counterpart to JMML with peripheral blood monocytosis, in both patients with germline and somatic ETV6 mutations.8 -10 These literature reports suggest that the novel ETV6 mutation affected megakaryocyte morphology in our case, potentially increasing the degree of dysplasia and the number of megakaryocytes beyond what is typically seen in JMML.
Another confounding factor seen in some cases of pediatric myeloid malignancy is a clinical presentation similar to other disorders, including benign disorders such as inherited immune deficiencies and autoimmune disorders. In our case, a clonal somatic KRAS G12D mutation was detected, which has been reported in association with RAS-associated autoimmune leukoproliferative disorder (RALD).3,14 The case had features that overlap with but are not specific for RALD: monocytosis, possible increase in CD4- CD8- T cells, recurrent infections, increased hemoglobin F, increased serum IgG, and increased serum vitamin B12. The case also had features that do not support RALD, including an absence of lymphadenopathy, an absence of B cell lymphocytosis, lack of evidence for autoimmunity, and multiple acquired somatic mutations. In particular, the presence of monosomy 7 and increased blasts necessitated a diagnosis of JMML in this context. Retrospective review of the case led us to consider whether the patient had RALD preceding the onset of JMML given the time course, history of an indeterminate immune deficiency, and the presence of CD4- CD8- T cells.
In summary, we report a case of JMML with unusually dysplastic megakaryocyte morphology which may be related to a novel ETV6 mutation in the setting of JMML, given the reports of ETV6 mutations associated with familial thrombocytopenia and various other myeloid malignancies. Additional studies will be needed to ascertain the role of ETV6 in JMML. This may also broaden our general understanding of the pathogenesis of JMML, which has changed over time and continues to evolve as related disorders such as RALD are discovered. The patient also had a prolonged clinical course prior to diagnosis, confounded by immune manifestations of uncertain etiology, which raised the possibility of RALD preceding JMML and underscored the need to establish definitive criteria for differentiating the two disorders.
Footnotes
Acknowledgements
The authors would like to thank Kristin Zajo, Certified Genetic Counselor at Nationwide Children’s Hospital, for her assistance in compiling and interpreting reference laboratory reports for this case.
Abbreviations
CBC, complete blood count; WBC, white blood cell count; hgb, hemoglobin; GM-CSF, granulocyte-macrophage colony-stimulating factor.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.