
Abstract
Salacia reticulata has long been used in traditional medicine for metabolic disorders, but clinical evidence for obesity-related outcomes remains limited. We conducted a 12-week randomized, double-blind, placebo-controlled trial to evaluate the preliminary efficacy and safety of a standardized Salacia reticulata extract (SLE) on body-fat outcomes. Adults with overweight and/or obesity were randomized to receive SLE capsules or a matching placebo twice daily for 12 weeks. The coprimary endpoints were changes from baseline to week 12 in total body fat mass (g) and body fat percentage (%) measured by dual-energy X-ray absorptiometry. The primary efficacy analysis was conducted in the full analysis set (mITT) using analysis of covariance with baseline values and sex as covariates. Per-protocol set analyses were performed as supportive sensitivity analyses. Among randomized participants, the mITT included 66 participants in the SLE group and 67 in the placebo group, and baseline characteristics were comparable. After 12 weeks, total body fat mass decreased in the SLE group, whereas little change was observed in the placebo group. The between-group difference favored SLE (ANCOVA-adjusted LS mean difference, −482.30 g; 95% CI, −907.05 to −57.54 g). The corresponding between-group comparison was statistically significant (Wilcoxon rank-sum test, P = .0158). Body fat percentage also decreased in the SLE group, but the between-group difference was not statistically significant (ANCOVA-adjusted LS mean difference, −0.30%; 95% CI, −0.73 to 0.12%; Wilcoxon rank-sum test, P = .1453). Several secondary body-composition outcomes, including trunk, android and gynoid fat measures, showed directionally favorable changes in the SLE group. In this 12-week trial, SLE was associated with a modest reduction in total body fat mass, whereas the second coprimary endpoint, body fat percentage, did not differ significantly between groups. These mixed coprimary findings provide only preliminary clinical evidence and should be interpreted with caution, particularly given the study’s retrospective trial registration.
Keywords
INTRODUCTION
Obesity is a complex metabolic disease driven by sustained energy intake exceeding energy expenditure and has become a major global health challenge. The global number of adults with overweight or obesity reached 2.11 billion in 2021.1,2 This rise is closely associated with lifestyle changes, including increased consumption of calorie-dense, nutrient-poor foods and reduced physical activity related to urbanization and sedentary work patterns. 3 Obesity predisposes individuals to insulin resistance, chronic low-grade inflammation in adipose tissue, and dysregulated adipocyte proliferation and differentiation, thereby increasing the risk of chronic diseases such as cardiovascular disease, type 2 diabetes mellitus, non-alcoholic fatty liver disease, and certain cancers. 4 It is also associated with osteoarthritis and generalized anxiety disorder.5,6
Alongside lifestyle modification and pharmacotherapy, nutraceutical approaches are increasingly considered because of their favorable safety profiles and sustainability.7,8 In particular, plant-derived nutraceuticals rich in phytochemicals may beneficially modulate metabolic pathways with relatively fewer adverse effects. 9 Several dietary supplements (e.g., Garcinia cambogia, alpha-lipoic acid, and vitamin D) have been investigated for obesity management, and related research remains ongoing.10–12
Among Salacia species, Salacia reticulata has a long history of use in Ayurvedic medicine in India and Sri Lanka for metabolic disorders, including diabetes and obesity. 13 Preclinical studies suggest that Salacia extracts may inhibit pancreatic lipase activity, suppress adipocyte differentiation, and improve insulin sensitivity.14–16 However, clinical evidence supporting efficacy and safety in human obesity remains limited; existing clinical studies have largely focused on type 2 diabetes 17 or indirect markers such as gastrointestinal peptide regulation. 18 Therefore, we conducted a randomized, double-blind, placebo-controlled trial to evaluate the preliminary efficacy and safety of a standardized Salacia reticulata extract (SLE) on body-fat outcomes in adults with overweight or obesity, using dual-energy X-ray absorptiometry (DEXA)-derived body-fat composition as the prespecified primary assessment. Given the limited prior clinical evidence, this trial should be regarded as exploratory, and its findings should be interpreted as preliminary rather than confirmatory evidence of efficacy.
MATERIALS AND METHODS
Study design and participants
This randomized, double-blind, placebo-controlled clinical trial was conducted at the Korean Medicine Hospital of Semyung University (Jecheon, Republic of Korea) in accordance with the Declaration of Helsinki and is reported in line with CONSORT guidelines. The definitions for all abbreviations and terms used throughout this study are provided in Supplementary Table S18. The protocol (version 1.0, dated November 9, 2022) was approved by the Institutional Review Board of Semyung University Korean Medicine Hospital (SMJOH-2022-11; approved December 2, 2022), and the trial was registered with the Korean Clinical Research Information Service (KCT0010387). Written informed consent was obtained from all participants. Participants were recruited via printed announcements on the hospital website and bulletin boards and screened in the internal medicine department according to eligibility criteria, as detailed in Table 1. Total participation lasted approximately 15 weeks, including up to a 3-week washout period and a 2-week follow-up after the final visit. Participants using prohibited medications or dietary supplements who had not discontinued them ≥14 days before screening completed a minimum 14-day washout, with consent obtained for discontinuation as needed. Randomization occurred at Visit 2 (within 3 weeks of Visit 1), which served as baseline, after reconfirming eligibility. Study products were dispensed for 33 days at baseline, and follow-up visits were scheduled on days 28, 56, and 84 (Visits 3–5; ±5-day window). At each visit, vital signs, medical history, concomitant medications, and efficacy/safety outcomes were assessed, as shown in Figure 1. Laboratory tests and pregnancy screening were performed at Visits 1 and 5.
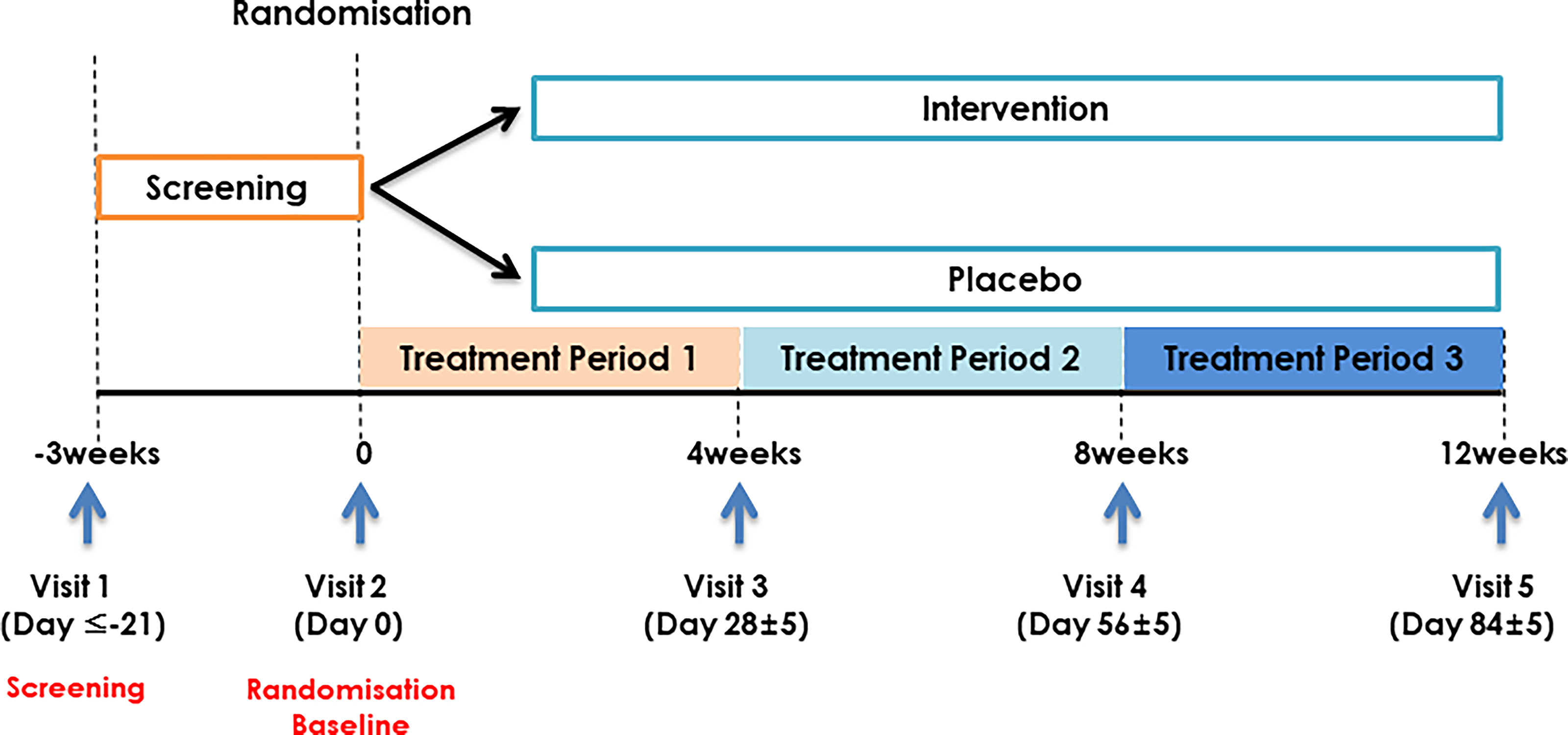
Clinical trial timeline.
Participant Selection Criteria

Participants must meet all of the inclusion criteria.
Participants with a stable history of cerebrovascular or cardiac disease could be included at the investigator’s discretion.
The classification of obesity in this study was based on the WHO Asia-Pacific BMI criteria. 19
AST, aspartate aminotransferase; ALT, alanine aminotransferase.
Intervention
Each active capsule (450 mg) contained 250.0 mg SLE, 74.0 mg crystalline cellulose, 112.5 mg maltodextrin, 4.5 mg magnesium stearate, and 9.0 mg silicon dioxide. Each placebo capsule (450 mg) contained 74.0 mg crystalline cellulose, 362.5 mg maltodextrin, 4.5 mg magnesium stearate, and 9.0 mg silicon dioxide, as detailed in Supplementary Table S1. Active and placebo capsules were indistinguishable in shape, size, and color, as shown in Figure 2.
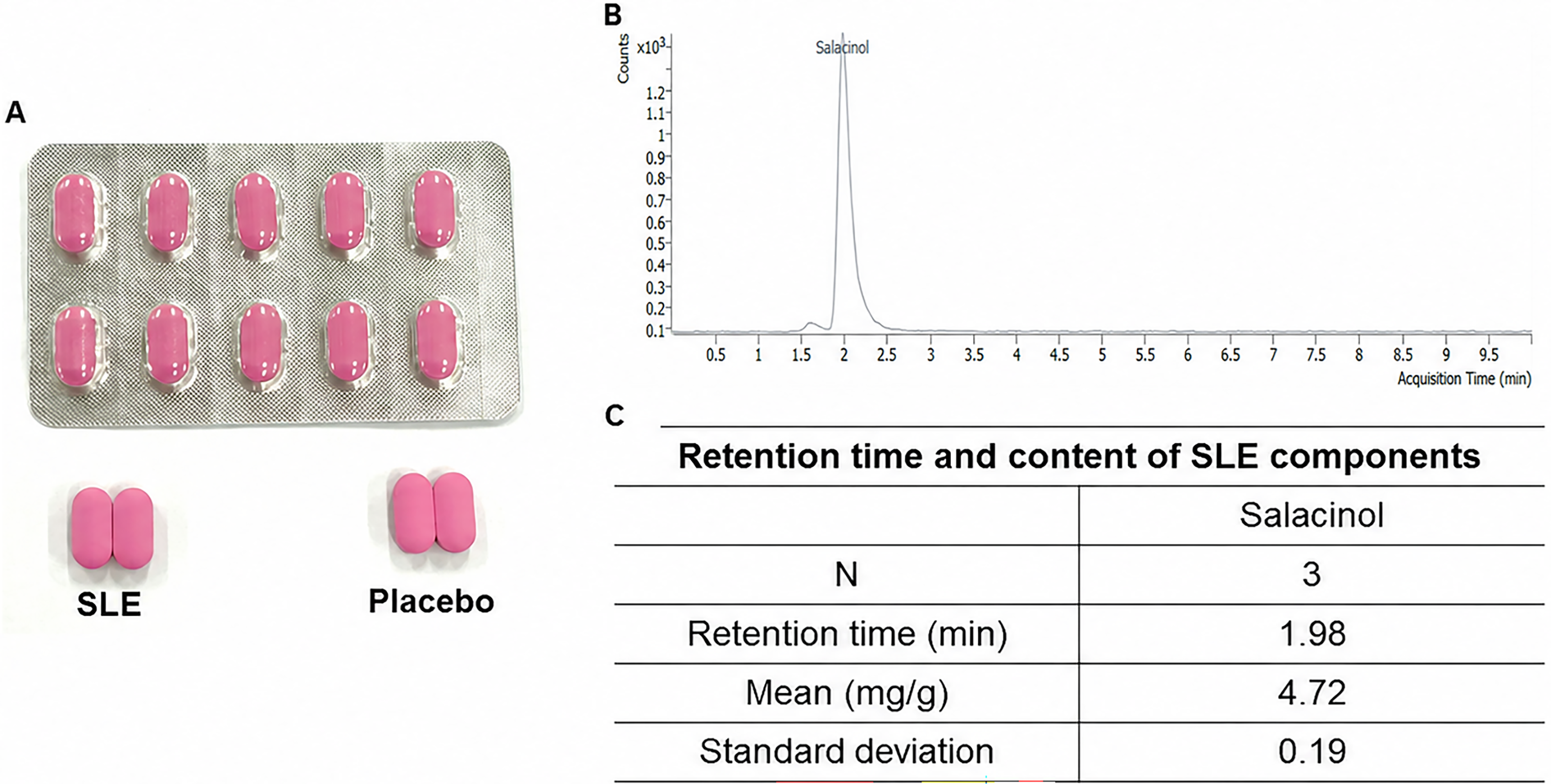
Study product appearance and chemical characterization of Salacia reticulata extract (SLE). (A) SLE and placebo capsules showing identical external appearance. (B) Representative liquid chromatography–mass spectrometry (LC–MS) chromatogram of SLE, with the salacinol peak identified at a retention time of approximately 1.98 min. (C) Retention time and salacinol content of SLE (n = 3; mean 4.72 ± 0.19 mg/g). SD, standard deviation.
The SLE was botanically authenticated and standardized by quantifying salacinol using liquid chromatography–mass spectrometry; thin-layer chromatography, and mass spectrometry were also performed to confirm batch consistency.
Participants took one capsule twice daily (before breakfast and dinner) for 12 weeks. At Visits 2–4, a 33-day supply was dispensed, unused capsules were returned, and compliance was assessed by capsule count.
Participants were instructed to maintain their usual diet and physical activity. Concomitant medications and other interventions potentially affecting body fat reduction were prohibited or restricted at the investigator’s discretion and were documented at each visit. The intervention was discontinued for serious adverse events, use of prohibited medications/procedures affecting body fat or lipid metabolism, withdrawal of consent, administrative reasons (e.g., noncompliance), or follow-up difficulties.
Randomization and blinding
Stratified block randomization was used to ensure balanced allocation and minimize bias. The randomization sequence was generated using SAS® (version 9.4; SAS Institute Inc., Cary, NC, USA) and stratified by sex at Visit 2. Randomization was implemented through a centralized interactive web response system (IWRS) with a reproducible seed. Prior to trial initiation, the sponsor labeled intervention packages according to investigational product (IP) code lists. An independent third party maintained the randomization codes and corresponding IP numbers, which remained concealed until completion of statistical analyses, except for medical emergencies. IP administrators (or pharmacists) dispensed study products according to IP numbers; if replacement was required (e.g., packaging issues or loss), the IWRS was used to maintain the original group assignment.
Participants, investigators, study coordinators, outcome assessors, and the statistician were blinded to treatment allocation. Active (SLE) and placebo capsules were identical in appearance, packaging, labeling, and dosing schedule, and treatment codes were inaccessible to the study team until database lock, except when emergency unblinding was necessary.
Primary and secondary endpoints
The coprimary endpoints were the changes from baseline to week 12 in total body fat mass (g) and body fat percentage (%) assessed by DEXA. Secondary endpoints included DEXA-derived body composition (lean mass and regional fat mass/percentage), CT-derived abdominal fat distribution, anthropometric indices, serum lipid profiles, adipokines, glucose-related parameters, physical activity, diet, and safety outcomes.
Total body fat mass and body composition were measured at baseline and at week 12 using whole-body DEXA (Lunar Prodigy Advance, GE Healthcare, Madison, WI, USA) with the manufacturer’s software; scans were reviewed by a blinded experienced operator. Abdominal adipose tissue was assessed at baseline and week 12 using multidetector CT (SOMATOM Definition AS+, Siemens Healthineers, Erlangen, Germany) with a single axial slice at L4–L5; adipose tissue was segmented using −190 to −30 Hounsfield units, and measurements were reviewed by a blinded radiologist.
Fasting (≥10–12 h) blood samples were collected at baseline and week 12. Serum lipids and glucose were analyzed using standard enzymatic colorimetric methods on an automated analyzer. Insulin, leptin, and adiponectin were measured using chemiluminescent immunoassay and/or commercial ELISA kits (Millipore, USA) per manufacturers’ instructions, and insulin resistance was estimated using HOMA-IR.
Safety assessment
Adverse events and safety concerns were monitored throughout the study. At each visit, participants were queried using open-ended and targeted questions, and events were recorded with type, severity, outcome, and relatedness to the study product. Adverse events were coded using MedDRA (version 26.0) and graded according to standard toxicity criteria.
Safety laboratory tests (hematology, serum chemistry, and urinalysis) were performed at baseline and week 12 using automated analyzers and standard enzymatic or colorimetric methods per manufacturers’ instructions. Assay kit details (product name, catalogue number, and supplier) are provided in Supplementary Table S2.
Sample-size calculation
Sample size was determined based on a previous study by Cho et al., 20 which reported a mean body fat mass reduction of −1.6 kg in the herbal extract group compared with −0.1 kg in the placebo group (P = .023). Using these data, the expected effect size was set at −1.5 kg, with an estimated pooled standard deviation of 2.27 kg. To achieve 93.5% statistical power at a two-sided 5% significance level, 56 participants per group were required. Assuming a 25% dropout rate, the final target sample size was 75 participants per group (150 total).
Statistical analysis
All analyses were two-tailed with a significance level of 0.05. Normality was assessed using the Shapiro–Wilk test. Continuous variables are presented as means ± SD (standard deviations) and were compared between groups using independent t-tests or Wilcoxon rank-sum tests, as appropriate; within-group changes were assessed using paired t-tests or Wilcoxon signed-rank tests. Categorical variables are presented as n (%) and were compared using Pearson’s chi-square or Fisher’s exact tests.
For normally distributed outcomes, treatment effects were evaluated using analysis of covariance (ANCOVA) with baseline values and sex as covariates. In this trial, the primary efficacy analysis was conducted in the full analysis set (FAS/mITT), defined as all randomized participants who received at least one dose of study product and had at least one post-baseline efficacy assessment. The per-protocol set (PPS), defined as participants who completed the trial without major protocol deviations, was analyzed as a supportive sensitivity population. For the mITT, missing post-baseline values were imputed using the last observation carried forward method; no imputation was applied for the PPS or SAS analyses. Prior to ANCOVA, the normality of variables was assessed using the Shapiro–Wilk test and visual inspection; when ANCOVA assumptions were not adequately met (e.g., due to outliers), the nonparametric Wilcoxon rank-sum test was applied as a prespecified fallback, with least-squares (LS) mean differences and 95% confidence intervals derived from the ANCOVA model for descriptive purposes. Multiplicity for the co-primary endpoints was controlled using a Bonferroni adjustment, allocating a two-sided α level of 0.025 to each endpoint. Statistical significance for the primary analysis required both co-primary endpoints to be significant. Key secondary endpoints were tested hierarchically and only if both coprimary endpoints achieved statistical significance; otherwise, secondary analyses were considered exploratory. The Bonferroni correction for co-primary endpoints and the hierarchical framework for key secondary endpoints were introduced as post-hoc analytical improvements during peer review and were not part of the original protocol (see Limitations). A separate formal multiplicity adjustment for all secondary endpoints was also not prespecified; accordingly, statistically significant secondary findings should be interpreted as exploratory. Safety analyses included all participants who received at least one dose and had at least one post-baseline safety assessment. All analyses were performed using SAS® (version 9.4; SAS Institute Inc., Cary, NC, USA). Statistical analyses were performed by RnBS Corp. (S.J.Y. and Y.C.) as an independent contracted statistical organization; the sponsor (
RESULTS
Participant characteristics
In total, 161 individuals were screened, of whom 11 did not meet the eligibility criteria. Ultimately, 150 participants were randomized into either the SLE group (n = 75) or the placebo group (n = 75). Of these, nine participants in the SLE group and eight in the placebo group discontinued the trial. Ultimately, 133 participants completed the study (66 in the SLE and 67 in the placebo group), as shown in Figure 3.
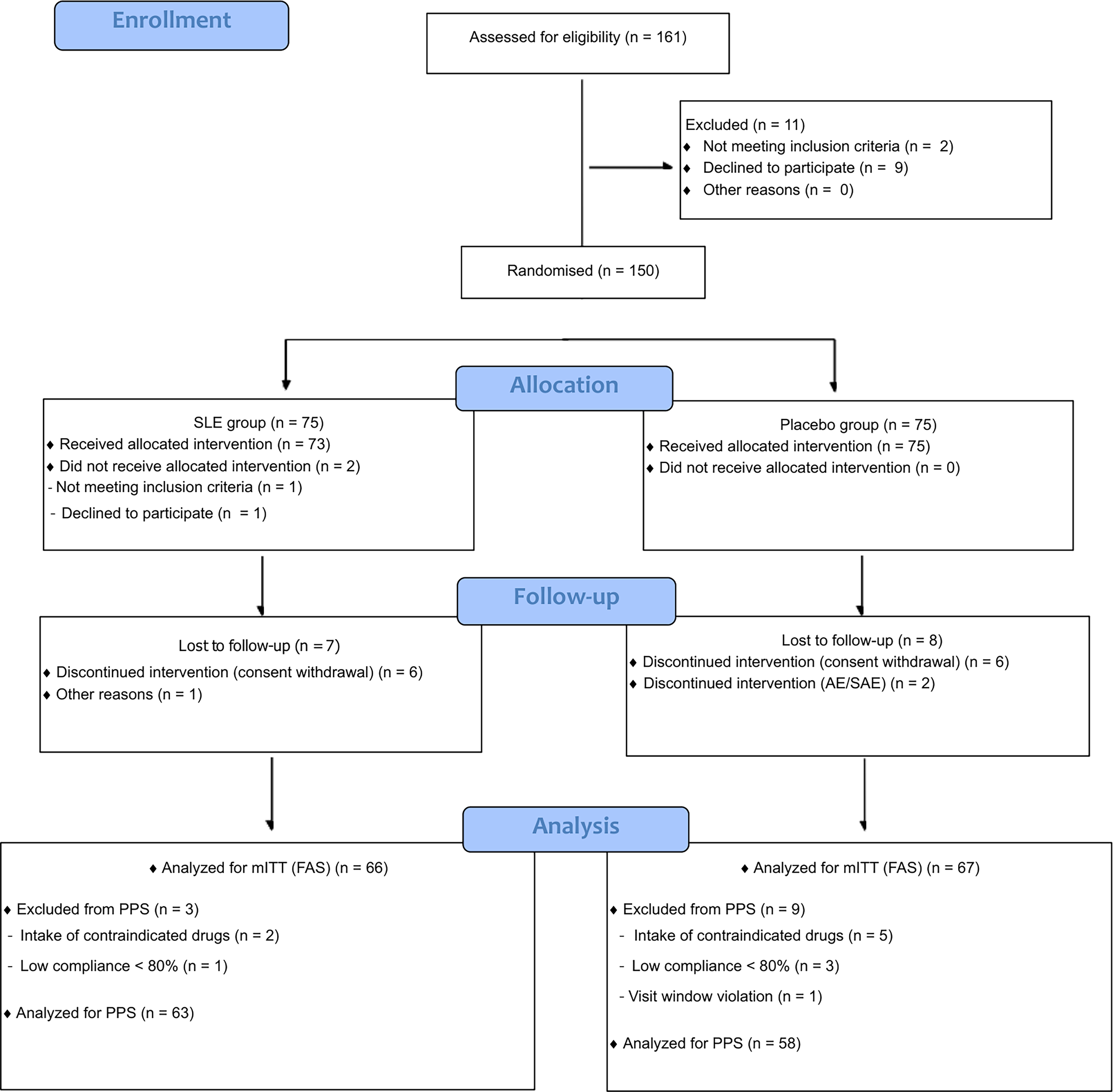
CONSORT flowchart. AE, adverse event; mITT, modified intention-to-treat/full analysis set; PPS, per-protocol set; SAE, serious adverse event; SLE, Salacia reticulata extract.
Baseline demographic characteristics showed no statistically significant differences between groups. The mean age was 44.27 ± 10.00 years in the SLE group and 44.40 ± 8.96 years in the placebo group (P = .8907). No significant differences were observed in sex distribution, height, or family history of obesity, as detailed in Table 2.
Participant Characteristics by Group (mITT)
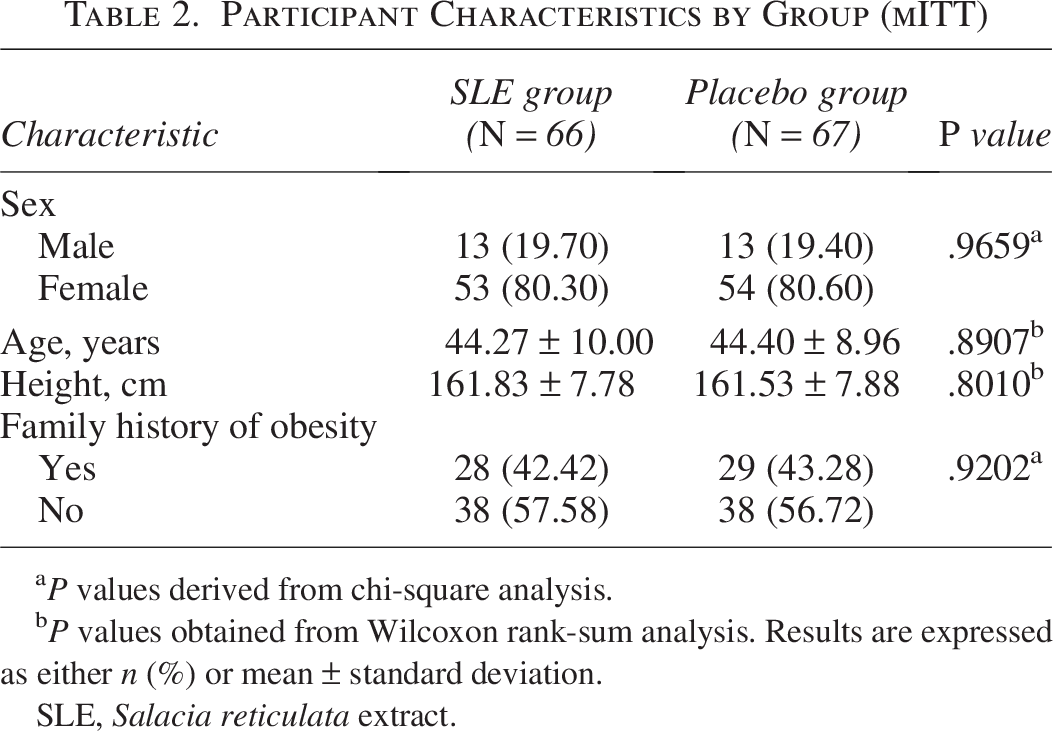
P values derived from chi-square analysis.
P values obtained from Wilcoxon rank-sum analysis. Results are expressed as either n (%) or mean ± standard deviation.
SLE, Salacia reticulata extract.
Study endpoints
In the primary mITT analysis (SLE, n = 66; placebo, n = 67), the coprimary findings were mixed. After 12 weeks of supplementation, total body fat mass decreased significantly from baseline in the SLE group (−582.52 ± 1,170.35 g, P < .0001), whereas no significant within-group change was observed in the placebo group (−17.73 ± 1,291.09 g, P = .9108). The between-group difference in total body fat mass was statistically significant (LS mean difference, −482.30 g; 95% CI, −907.05 to −57.54 g; P = .0158, Wilcoxon rank-sum test; Table 3). Body fat percentage also decreased within the SLE group (−0.44 ± 1.26%, P = .0117), whereas no significant within-group change was observed in the placebo group (−0.06 ± 1.26%, P = .7129). However, the between-group difference in body fat percentage was not statistically significant (LS mean difference, −0.30%; 95% CI, −0.73 to 0.12%; P = .1453, Wilcoxon rank-sum test; Table 3). Accordingly, the coprimary results should be interpreted as mixed and exploratory rather than confirmatory. Figure 4 presents representative DEXA images for illustrative purposes only.
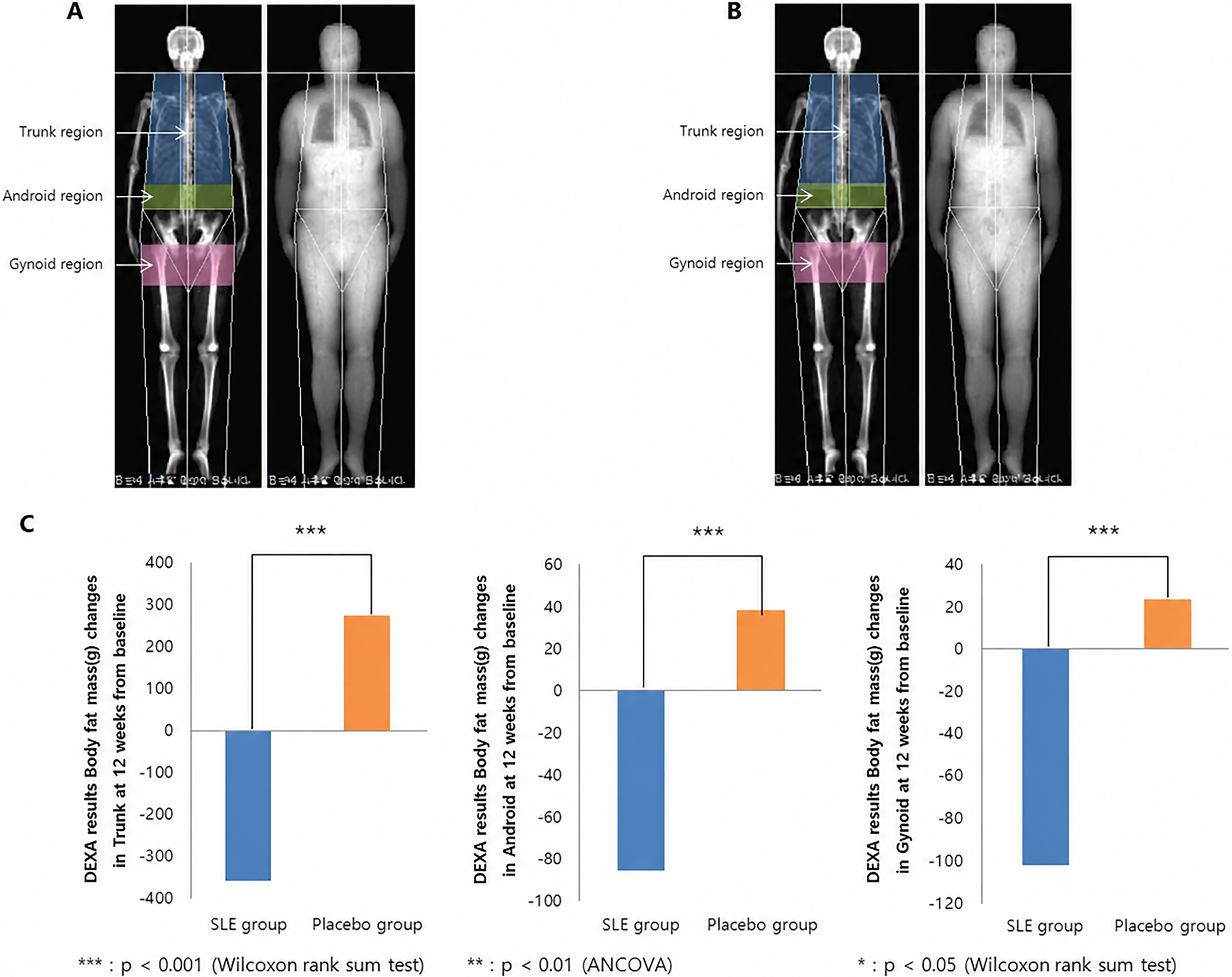
Dual-energy X-ray absorptiometry (DEXA) images before and after participation in the intervention group.
Dual-Energy X-Ray Absorptiometry (DEXA) Measurements at Week 12 Compared with Baseline (mITT)

P < .05 *, P < .01 **, P < .001 ***.
Wilcoxon signed-rank test P value.
Paired t-test P value.
ANCOVA analysis adjusted for baseline measurements and sex.
Wilcoxon rank-sum test P value.
ANCOVA P value adjusted for baseline and sex.
Values are expressed as mean ± standard deviation.
ANCOVA, analysis of covariance; Px, placebo group; V2, Visit 2; V5, Visit 5; LS, least-squares; CI, confidence interval; Tx, treatment (SLE) group.
Among the secondary endpoints, several DEXA-derived regional body-composition outcomes, including trunk, android and gynoid fat measures, showed directionally favorable changes in the SLE group (Supplementary Table S4). Body weight and BMI also showed greater reductions in the SLE group (Supplementary Table S5). Modest changes in waist and hip circumference were additionally observed, although between-group differences were limited (Supplementary Table S6). These secondary findings should be regarded as exploratory.
CT-based abdominal fat outcomes did not show consistent between-group superiority for SLE. Subcutaneous adipose tissue area decreased in the SLE group (−7.86 ± 49.31 cm2) but increased in the placebo group (+4.98 ± 33.72 cm2), with an LS mean between-group difference of −7.87 cm2 (95% CI: −20.74 to 5.00; P = .2285 ANCOVA). Visceral adipose tissue area showed minimal within-group changes in both groups (SLE: +0.17 ± 28.10 cm2; placebo: −3.53 ± 22.39 cm2), with no significant between-group difference (LS mean difference, + 4.48 cm2; 95% CI: −3.76 to 12.72; P = .2060, Wilcoxon rank-sum test). In particular, the visceral-to-subcutaneous fat area ratio showed a significantly greater reduction in the placebo group than in the SLE group at week 12 (LS mean difference, + 0.05; 95% CI: −0.02 to 0.12; P = .0266, Wilcoxon rank-sum test; Supplementary Table S7). Figure 5 shows representative CT scans from one participant in the SLE group for illustrative purposes only and should not be interpreted as group-level evidence of efficacy. Other secondary endpoints showed no statistically significant changes in either group.
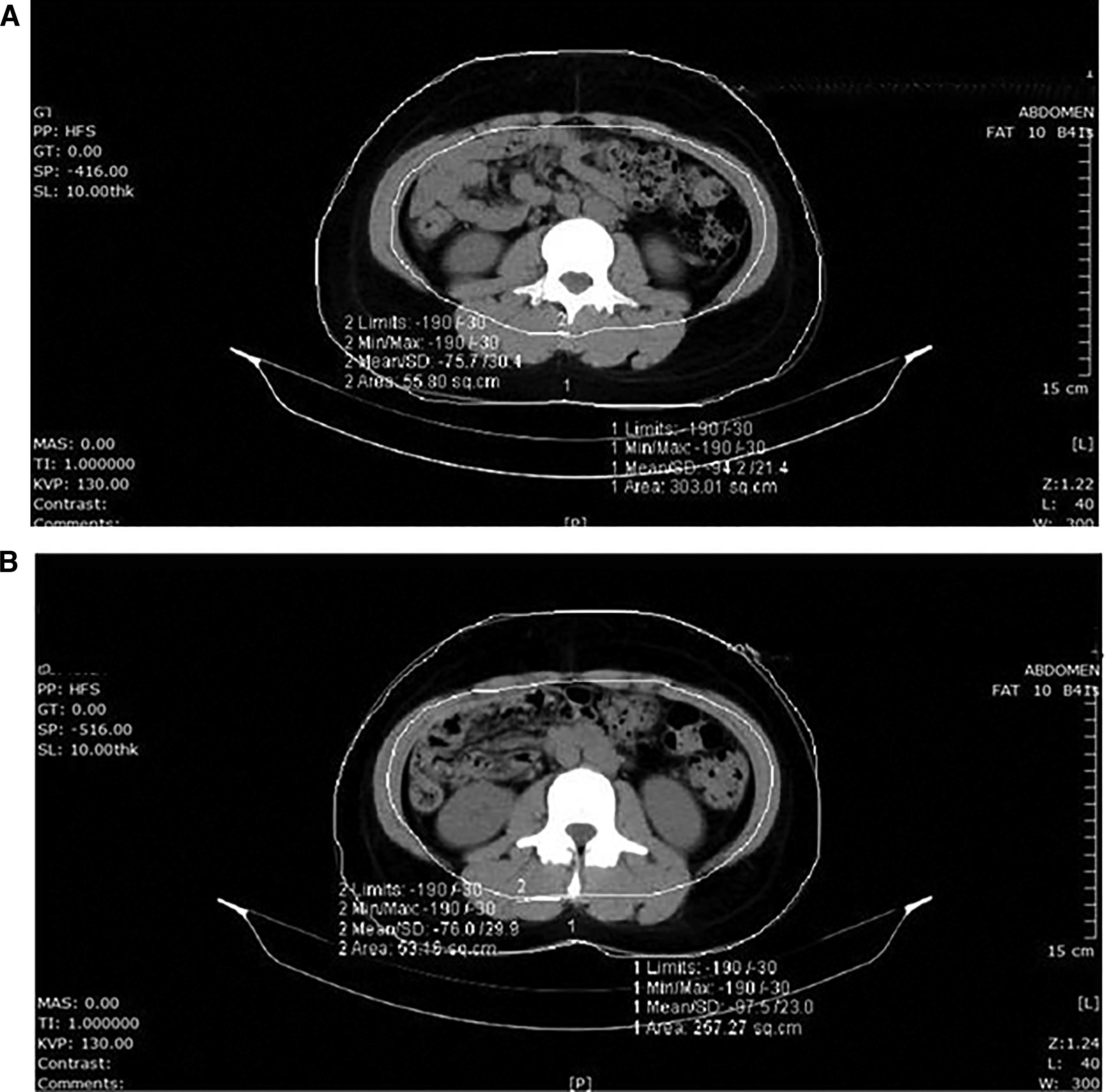
Computed tomography (CT) images before and after participation in the intervention group. Axial CT slices at the L4-L5 intervertebral level at
Serum lipid concentrations, adipokines, fasting glucose, fasting insulin, and HOMA-IR did not show statistically significant between-group differences (Supplementary Table S8).
Supportive per-protocol analysis
In the supportive per-protocol (PPS) analysis (SLE, n = 63; placebo, n = 58), the direction and magnitude of treatment effects were consistent with the primary mITT results. Total body fat mass showed a statistically significant between-group difference favoring SLE (LS mean difference, −549.38 g; 95% CI: −971.22 to −127.53; P = .0073, Wilcoxon rank-sum test; Supplementary Table S3), while body fat percentage did not differ significantly between groups (LS mean difference, −0.35%; 95% CI: −0.78 to 0.08; P = .1258, Wilcoxon rank-sum test). The consistency between the PPS and mITT results supports the robustness of the primary outcome. PPS results for secondary body-composition, anthropometric, CT-based, and metabolic endpoints are presented in Supplementary Tables S13, S14, S15, S16, and S17.
Safety
In the SLE group, 59 adverse events were reported among 32 participants (43.84%), while 52 AEs occurred among 34 participants (45.33%) in the placebo group, with no significant differences between groups (P = .8546 Pearson’s chi-square test). Two serious AEs occurred in the placebo group—one hospitalization due to a traffic accident and another due to acute pancreatitis; both participants fully recovered after discharge. All AEs were mild in intensity and deemed unrelated to the intervention, as detailed in Supplementary Table S9.
For hematological parameters, some statistically significant within-group changes were observed in the SLE group after 12 weeks; however, all remained within normal ranges and were not clinically meaningful. Between-group differences were not significant (Supplementary Table S10). Similarly, albumin and insulin levels showed significant within-group changes from baseline in the SLE group but remained within normal limits, and no significant between-group differences were observed. No clinically meaningful changes were detected in other hematological, biochemical, or urinalysis parameters, as detailed in Supplementary Tables S10 and S11.
Physical activity and diet
No statistically significant differences were found between the two groups for physical activity levels or energy expenditure at baseline (Visit 2), after 12 weeks of intake (Visit 5), or in changes during the study period, as detailed in Supplementary Table S12.
DISCUSSION
In this randomized trial, SLE was associated with a statistically significant reduction in total body fat mass compared with placebo after 12 weeks. However, the second co-primary endpoint, body fat percentage, was not significantly different between groups. Accordingly, the efficacy findings should be interpreted as mixed and preliminary rather than confirmatory.
Among the secondary outcomes, reductions in trunk, android, and gynoid fat and modest decreases in body weight and BMI were observed in the SLE group. Specifically, the between-group difference in body weight was −0.83 kg (95% CI: −1.41 to −0.24) and in BMI was −0.31 kg/m2 (95% CI: −0.53 to −0.09); while statistically significant, these changes are modest in absolute magnitude and of uncertain clinical relevance. These findings may indicate a short-term signal toward reduced adiposity, particularly in regional fat mass; however, the magnitude of change was modest, and these secondary results should be interpreted cautiously because no formal multiplicity adjustment was applied.
CT-derived abdominal fat findings were not fully consistent with a treatment effect. In particular, the visceral-to-subcutaneous fat area ratio improved more in the placebo group than in the SLE group. Accordingly, the CT findings do not provide clear evidence of efficacy and should be interpreted only as exploratory and hypothesis-generating.
Preclinical studies suggest several biologically plausible mechanisms for Salacia reticulata, including effects on pancreatic lipase activity, adipogenesis-related pathways, and AMPK signaling. However, these mechanisms were not directly assessed in the present clinical trial, and no mechanistic inference can be drawn from the current human data. The proposed pathways should therefore be regarded as biologically plausible background rather than demonstrated clinical mechanisms.
This study has several important limitations. First, the trial was registered retrospectively, which limits confidence regarding the prospective specification of endpoints and analysis strategy. Second, the original protocol designated the PPS as the primary efficacy analysis and did not specify a formal multiplicity correction for coprimary endpoints or a hierarchical testing procedure for secondary endpoints. The revision of the primary analysis to the mITT, the addition of the Bonferroni correction (α = 0.025 per coprimary endpoint), and the hierarchical framework for secondary endpoints were introduced during peer review as post-hoc methodological improvements and were not prospectively prespecified. These analytical changes strengthen the interpretive framework but should be recognized as retrospective revisions. Because the two coprimary endpoints yielded mixed results and multiple secondary outcomes were analyzed without a prospectively prespecified formal multiplicity adjustment, statistically significant secondary findings should be regarded as exploratory rather than confirmatory. Third, the 12-week duration, single-center setting, and absence of direct mechanistic measurements further limit the generalizability and interpretation of the findings.
No clinically meaningful safety concerns were identified over 12 weeks, broadly consistent with prior reports.9,17,21,22 Longer-term safety requires further evaluation.
CONCLUSIONS
In this 12-week randomized, double-blind, placebo-controlled trial, SLE was associated with a modest reduction in total body fat mass, whereas the second coprimary endpoint, body fat percentage, did not differ significantly between groups. These mixed coprimary findings provide only preliminary clinical evidence and should be interpreted cautiously, particularly in light of the retrospective trial registration and post-hoc analytical revisions. Larger, prospectively registered trials with clearly prespecified endpoints, estimands, and multiplicity control are needed to confirm clinical efficacy.
AUTHORS’ CONTRIBUTIONS
Conceptualization, S.M.S. and Y.-J.C.; methodology, S.-H.P., J.-E.L., S.-G.B., S.J.Y., Y.C., and S.M.S.; formal analysis, S.J.Y. and Y.C.; investigation, S.-H.P., J.-E.L., and S.-G.B.; resources, J.K., K.-S.B., and Y.G.; data curation, S.J.Y. and Y.C.; writing—original draft preparation, S.-H.P. and S.M.S.; writing—review and editing, all authors; supervision, S.M.S.; project administration, S.M.S.; funding acquisition, J.K., K.-S.B., Y.G., and S.M.S. All authors have read and agreed to the published version of the article.
AVAILABILITY OF DATA AND MATERIALS
The datasets supporting the conclusions of this article are included within the article and its additional file.
Footnotes
ACKNOWLEDGMENTS
The authors thank Professor Heung Ko and Seung-Min Lee for their valuable advice and support during study planning and article preparation.
AUTHOR DISCLOSURE STATEMENT
S.H.P., J.E.L., S.G.B., and S.M.S. are affiliated with Semyung University. S.J.Y. and Y.C. are employed by RnBS, which provided trial operational and statistical support. J.K., K.S.B., and Y.G. are employed by
FUNDING INFORMATION
This study was funded by
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.