
Abstract
Excessive lipid accumulation is a hallmark of metabolic disorders which includes obesity and insulin resistance; however, effective therapeutic strategies remain limited. Tamarixetin (Tx), a naturally occurring flavonoid with diverse pharmacological properties, has not been fully characterized in the context of lipid metabolism. In this study, we explored the metabolic benefits and molecular mechanisms of Tx in a Western diet (WD)-induced obesity model. Transcriptomic profiling revealed that Tx reversed WD-induced gene expression patterns, notably suppressing Pdk4 and inducing Phlda1 expression. Mechanistically, docking analysis suggested that Tx interacts with the acetyl-CoA-binding region within the p300 histone acetyltransferase domain, thereby attenuating H3K9 acetylation at the Pdk4 promoter. This epigenetic inhibition of Pdk4 led to activation of the p38/AMPK signaling cascade, upregulation of PPARGC1A and CPT1A, and enhanced insulin sensitivity in vitro. Collectively, our findings identify Tx as a novel epigenetic modulator that simultaneously suppresses lipogenic gene expression and restores metabolic signaling. Given its natural origin and multifaceted mode of action, Tx emerges as a promising candidate for therapeutic intervention in metabolic disorders.
INTRODUCTION
Excessive lipid accumulation is a contributing factor to various metabolic disorders, including obesity, type 2 diabetes (T2D), hyperlipidemia, non-alcoholic fatty liver disease (NAFLD), and cardiovascular disorders.1,2 These conditions primarily arise from dysregulated metabolic processes 3 and are often influenced by dietary factors, physical activity levels, and genetic predisposition.4–6 Between 2000 and 2019, the global prevalence of metabolic diseases increased significantly, with the largest increase observed in countries with high sociodemographic indices. 7 Because these disorders are interrelated, with the presence of one increasing the risk of another, comprehensive management and prevention strategies are essential. However, despite extensive research, a comprehensive understanding of the underlying molecular mechanisms remains elusive.
Pyruvate dehydrogenase kinase 4 (PDK4) is a key regulator of pyruvate dehydrogenase (PDH), which functions as a metabolic control point that balances glucose and fatty acid oxidation. 8 By inhibiting PDH activity, PDK4 promotes the influx of acetyl-CoA from β-oxidation into the tricarboxylic acid cycle, thereby altering energy metabolism. 9 Dysregulated PDK4 expression has been closely linked to metabolic dysfunction, particularly insulin resistance, obesity, and T2D.10–12 Recent findings suggest that aberrant PDK4 expression is also associated with heart failure and cardiovascular disease.13,14 Furthermore, because PDK4 plays a central role in glucose metabolism and mitochondrial respiration, its inhibition has been proposed as a novel therapeutic strategy for cancer. 15 Although previous studies have established a correlation between dysregulated PDK4 expression and metabolic disease pathogenesis, the epigenetic and transcriptional mechanisms regulating PDK4 expression remain insufficiently explored.
Recent studies have highlighted the importance of epigenetic regulation in metabolic homeostasis, particularly through histone modifications such as acetylation.16,17 Histone acetyltransferases (HATs), including p300, regulate chromatin accessibility and the transcriptional activation of metabolic genes involved in glucose and lipid metabolism. 18 In particular, p300-mediated histone acetylation has been implicated in the transcriptional regulation of genes associated with mitochondrial metabolism and lipid homeostasis, including Pdk4.19,20 These findings suggest that epigenetic regulation may represent an important mechanism underlying metabolic dysfunction and its therapeutic modulation.
Natural bioactive compounds derived from dietary sources have been extensively studied because of their stability and diverse pharmacological properties.21,22 These compounds exhibit anti-inflammatory, antioxidant, antimicrobial, hypoglycemic, anti-hypertensive, neuroprotective, cardioprotective, and hepatoprotective effects. Consequently, employing natural bioactive compounds as adjunctive therapies may serve as effective therapeutic and preventive strategies to mitigate the development and progression of various metabolic diseases. Tamarixetin (Tx), a flavonoid derivative of quercetin, is abundant in various plants and has demonstrated cardioprotective, 23 renoprotective, 24 anti-inflammatory, 25 antioxidant, 26 and antitumor 27 properties. In addition, flavonoids and quercetin derivatives have been reported to regulate metabolic signaling pathways and epigenetic modifications associated with lipid metabolism and insulin sensitivity. However, despite these pharmacological benefits, the potential of Tx to regulate lipid metabolism and its specific role in modulating PDK4 have not been investigated.
To address this gap, the present study aimed to investigate the antilipogenic effects of Tx and the underlying molecular mechanisms. Specifically, we examined the in vivo impact of Tx on lipid metabolism and conducted RNA sequencing to identify PDK4 as a novel regulatory target of Tx. Understanding the interaction between Tx and PDK4 may provide new insights into its therapeutic potential for metabolic diseases.
MATERIALS AND METHODS
Cell culture and reagents
HepG2 cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in high-glucose Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum and antibiotics (Welgene, Daegu, Korea). Cells were maintained at 37°C in a humidified incubator with 5% CO2. To induce lipogenesis, HepG2 cells were treated with a nonfat bovine serum albumin (BSA)-conjugated mixture of oleic acid and palmitic acid (OPA; a mixture of 600 μM oleic acid and 150 μM palmitic acid) at a 4:1 ratio (Sigma-Aldrich, St. Louis, MO, USA). Tx was purchased from Sigma-Aldrich and administered concurrently with OPA at the indicated concentrations (3–100 μM) for 18 h to assess its effects on hepatic lipid accumulation. Sodium dichloroacetate (DCA, 2.5–10 mM), a selective PDK4 inhibitor, was obtained from Selleck Chem (Houston, TX, USA).
Animal experiments
Five-week-old male C57BL/6 mice were procured from Nara Biotech (Seoul, Korea) and housed under standardized conditions with a 12-hour light/dark cycle. All procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Korea Food Research Institute (KFRI-M-19005). Following a 1-week acclimation period, mice were randomly assigned to experimental groups (n = 5 per group) and fed either a control diet (CD) or a Western diet (WD). The WD used in this study was the commercially available D12079B diet (Research Diets Inc., NJ, USA), which is widely used to induce obesity and metabolic dysfunction in rodent models. The CD consisted of the Teklad Irradiated Global 18% Protein Rodent Diet (2918C; Inotiv, USA). Detailed nutritional compositions of both diets are provided in Supplementary Table S1. To assess the effects of Tx, Tx was uniformly mixed into the WD at concentrations of 0.01% or 0.025% (w/w), and mice were continuously fed the respective Tx-containing diets throughout the experimental period. The selected concentrations of Tx were determined based on previous studies investigating dietary flavonoids and quercetin derivatives, as well as preliminary dose-finding experiments conducted to ensure biological efficacy without overt toxicity.28–30 The Tx-containing diets were freshly prepared and provided ad libitum. Accordingly, mice were divided into four groups: (1) CD, (2) WD, (3) WD + 0.01% Tx, and (4) WD + 0.025% Tx. Mice were maintained on their respective diets for 12 weeks, and body weight was recorded weekly. At the end of the experimental period, mice were anesthetized with isoflurane, and blood samples were collected for serum isolation. Liver tissues were immediately harvested, weighed, and processed for subsequent analyses.
Quantitative real-time polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and used as a template for complementary DNA (cDNA) synthesis. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis was performed using the iCycler iQ system (Bio-Rad, Hercules, CA, USA) and SYBR Green PCR master mix (Thermo Fisher Scientific, Waltham, MA, USA). PCR amplification was conducted using primer sets listed in Supplementary Table S2. β-actin (Actb) mRNA was used for normalization.
Transcriptome analysis
To evaluate the impact of dietary intervention on gene expression, mRNA sequencing was performed using the Illumina NovaSeq 6000 system. Nine mice were divided into three groups (CD, WD, and WD with 0.025% Tx), with three mice per group. Sequencing reads were mapped to the mouse reference genome (mm10/GRCm38), generating read count files for each sample. The data were analyzed using iDEP 2.0 (https://bioinformatics.sdstate.edu/idep), 31 which was used for preprocessing, normalization, differential gene expression (DEG) analysis, and data visualization. DEGs were defined as genes with an absolute log2 fold change (|log2FC|) > 1 and an adjusted P value <.05.
Immunoblot assay
Cells were lysed in RIPA buffer (Elpis, Daejeon, Korea) supplemented with protease and phosphatase inhibitors (Roche, Basel, Switzerland) and incubated on ice for 30 min. Lysates were centrifuged at 20,000g for 20 min at 4°C. The supernatants were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the proteins were transferred onto nitrocellulose membranes (Bio-Rad Laboratories). The membranes were blocked with 5% DifcoTM nonfat skim milk solution for 30 min before being incubated with primary antibodies (listed in Supplementary Table S3). After washing, the membranes were incubated with secondary antibodies (Thermo Fisher Scientific), and protein bands were detected using the FUSION Solo S (Vilber Lourmat, Zac de Lamirault, France).
Chromatin immunoprecipitation assay
Liver tissues from mice were used for chromatin immunoprecipitation (ChIP) assays. Samples were fixed with 1% formaldehyde for 10 min, and cross-linking was quenched using 125 mM glycine for 5 min at 25°C. Subsequent steps followed the Pierce Agarose ChIP Kit (Thermo Fisher Scientific) protocol. ChIP was performed using H3K9ac and H3K14ac antibodies (Cell Signaling), with SDS excluded from all buffers. The primer sequences used for ChIP-qPCR are listed in Supplementary Table S2.
Nuclear fractionation from liver tissues
Liver tissues were collected at the end of the animal experiments and processed using the nuclear fractionation protocol provided by the manufacturer (Abcam, Cambridge, MA, USA). Briefly, tissues were weighed, finely minced, and homogenized in pre-extraction buffer (Thomas Scientific, NJ, USA) using a Teflon pestle (Thomas Scientific). The homogenates were centrifuged at 18,800 g for 15 min at 4°C, and the supernatants were collected as cytosolic fractions. The remaining pellets were resuspended in extraction buffer, homogenized, and centrifuged again under the same conditions. The resulting supernatants were collected as nuclear fractions.
Histone acetyltransferase activity assay
Mouse liver nuclear extracts (BioVision Biotechnology, Milpitas, CA, USA) were used as a source of histone acetyltransferase (HAT) enzymes. HAT activity in the nuclear extracts was assessed using a commercially available HAT activity assay kit, following the manufacturer’s protocol (BioVision Biotechnology).
Histone extraction
HepG2 cells were seeded at a density of 5 × 106 cells per 10-mm dish. When the cells reached ∼70% confluence (∼2 × 108), they were treated with OPA in the presence or absence of Tx for 18 h. Histones were extracted according to the manufacturer’s protocol (Abcam, Cambridge, MA, USA). Briefly, cells were lysed using a pre-lysis buffer to isolate the nuclear fraction. The nuclear pellet was resuspended in lysis buffer, incubated on ice for 30 min, and centrifuged at 9,600 g for 5 min at 4°C. The resulting supernatant was transferred to a clean tube, and DTT buffer was added for downstream analysis.
Cell viability assay
HepG2 cells were seeded into 24-well plates at a density of 5 × 104 cells/well. Upon reaching ∼70% confluence, the cells were treated with Tx at specified concentrations, with or without OPA. After 24 h of incubation, cell viability was assessed using a water-soluble tetrazolium salt (WST-1) solution. Briefly, 10 µM of WST-1 solution (Enzo Life Sciences, Inc., Farmingdale, NY, USA) was added to each well, and the plates were incubated for 3 h. Following incubation, 100 µL of the supernatant was transferred to a 96-well plate, and absorbance was measured at 450 nm using a spectrophotometer (Molecular Devices, Sunnyvale, CA, USA).
Docking simulation of Tx and the p300 HAT domain
Docking simulations were performed to assess the binding interactions between Tx and the p300 HAT domain. The crystal structure of the p300 HAT domain complexed with the bi-substrate inhibitor Lys-CoA (PDB ID: 3BIY) 32 was obtained from the Protein Data Bank and used as the protein structure for docking. The Tx ligand structure was generated using the following SMILES notation: COC1 = C(C = C(C = C1)C2 = C(C(=O)C3 = C(C = C(C = C3O2)O)O)O)O. Docking simulations were performed using the CB-Dock2 web server (https://cadd.labshare.cn/cb-dock2/index.php). 33 The p300 HAT domain structure (PDB ID: 3BIY) and the generated Tx structure were uploaded to Cb-Dock2, which predicted the optimal binding pose and binding affinity of Tx within the active site of p300.
Hematoxylin and eosin staining
Liver specimens were fixed in 4% formalin, embedded in paraffin, and sectioned into 4–5-µm-thick slices. The sections were then stained with hematoxylin and eosin (H&E) following standard histological procedures. Lipid accumulation in liver tissue was examined microscopically using an Eclipse 80i microscope (Nikon Instruments, Inc., Melville, NY, USA).
Measurements of serum glucose, cholesterol, and liver enzymes
Serum glucose, cholesterol, glutamic pyruvic transaminase, and glutamic oxaloacetic transaminase levels were measured using an enzymatic assay kit (Asan Pharm, Seoul, Korea) according to the manufacturer’s protocol.
Statistical analysis
All data are presented as the mean ± standard error of the mean (SEM), unless otherwise specified. Data in Figures 4 and 6 are presented as mean ± standard deviation (SD). Statistical comparisons between experimental groups were performed using one-way analysis of variance, followed by Tukey’s or Dunnett’s multiple-comparisons test, as appropriate, to identify pairwise differences. Statistical significance was defined as P < .05. For the graphical representation of group-wise differences, group letters were assigned to each group based on Tukey’s or Dunnett’s post hoc test. Groups not sharing a common letter were considered significantly different. All statistical analyses and visualizations were performed using GraphPad Prism 9 software (GraphPad Prism version 9.0.2, GraphPad Software, Boston, MA, USA).
RESULTS
Tx modulates gene expression in response to WD supplementation
In a previous study, we demonstrated the antilipogenic effects of Tx in 3T3-L1 preadipocytes. To identify novel targets involved in Tx-mediated metabolic regulation, we conducted a transcriptomic analysis of liver tissues from mice fed a CD, a WD, or WD + Tx diet. Heatmap visualization revealed distinct gene expression patterns across the dietary groups (Fig. 1A). A total of 132 genes were upregulated in the WD group compared with the CD group, whereas 37 genes were downregulated in the WD + Tx-fed group relative to the WD group (Fig. 1B, left panel). In contrast, 110 genes were downregulated in response to WD supplementation, whereas 40 genes exhibited increased expression following WD + Tx intake (Fig. 1B, right panel). DEGs were identified as described in the “Materials and Methods” section (|log2 fold change| > 1, adjusted P value < .05). Among the DEGs, five genes (913040I23Rik, Pdk4, Npnt, Cfd, and Phospho1) exhibited increased expression following WD supplementation, which was reversed by Tx administration. In contrast, five genes (Cabyr, Gnat1, Phlda1, Ppp1r10, and Gm7694) that were downregulated in the WD group exhibited restored expression following Tx intake (Fig. 1C). To confirm whether these changes in gene expression were consistent in liver tissues, we performed qRT-PCR analysis of Pdk4, Cfd, Phlda1, Ppp1r10, Gm7694, and Cabyr expression (Fig. 1D). Consistent with the DEG analysis, WD-induced Pdk4 and Cfd expression decreased following Tx supplementation. Conversely, Phlda1, Ppplr10, and Gm7694, which were downregulated in the WD group, were upregulated upon Tx administration. Notably, Cabyr expression in mouse liver tissues did not align with the DEG analysis results, exhibiting a different expression pattern (Supplementary Fig. S1). These findings suggest that Tx modulates the expression of specific metabolic genes in liver tissues through epigenetic regulatory mechanisms.
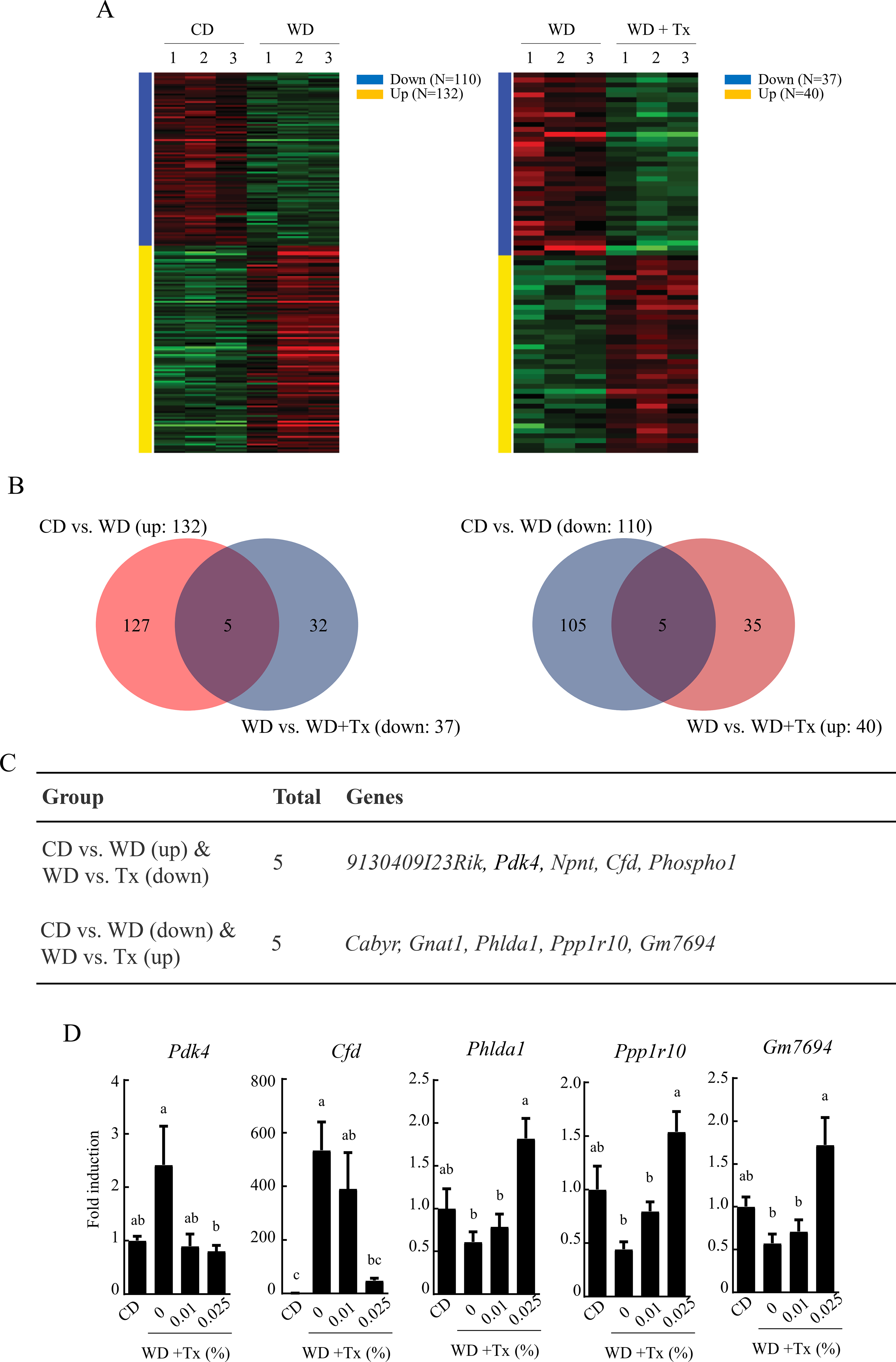
Transcriptomic analysis of the effects of tamarixetin (Tx) in a Western diet-induced obesity mouse model.
Tx inhibits histone H3K9 acetylation by suppressing HAT activity
Histone acetylation is a key regulatory mechanism controlling metabolic gene expression. To determine whether Tx modulates HAT activity, we measured HAT enzymatic activity in liver tissues. As shown in Figure 2A, HAT activity was significantly increased in the WD-fed group but was suppressed in the WD + Tx group. To examine whether alterations in HAT activity influenced histone acetylation, we examined histone H3 acetylation, a key marker of transcriptional activation in metabolic regulation (Fig. 2B). Histone H3K9 acetylation levels tended to increase in the livers of WD-fed mice, whereas this elevation appeared to be attenuated following Tx intake. However, this inhibition was not dose dependent. These results suggest that Tx suppresses histone H3K9 acetylation by inhibiting WD-induced HAT activity, thereby modulating the transcription of metabolism-related genes. Given that p300 is a major HAT associated with H3K9 acetylation and metabolic gene regulation, these findings support the rationale for further investigating the role of p300 in Tx-mediated epigenetic regulation.
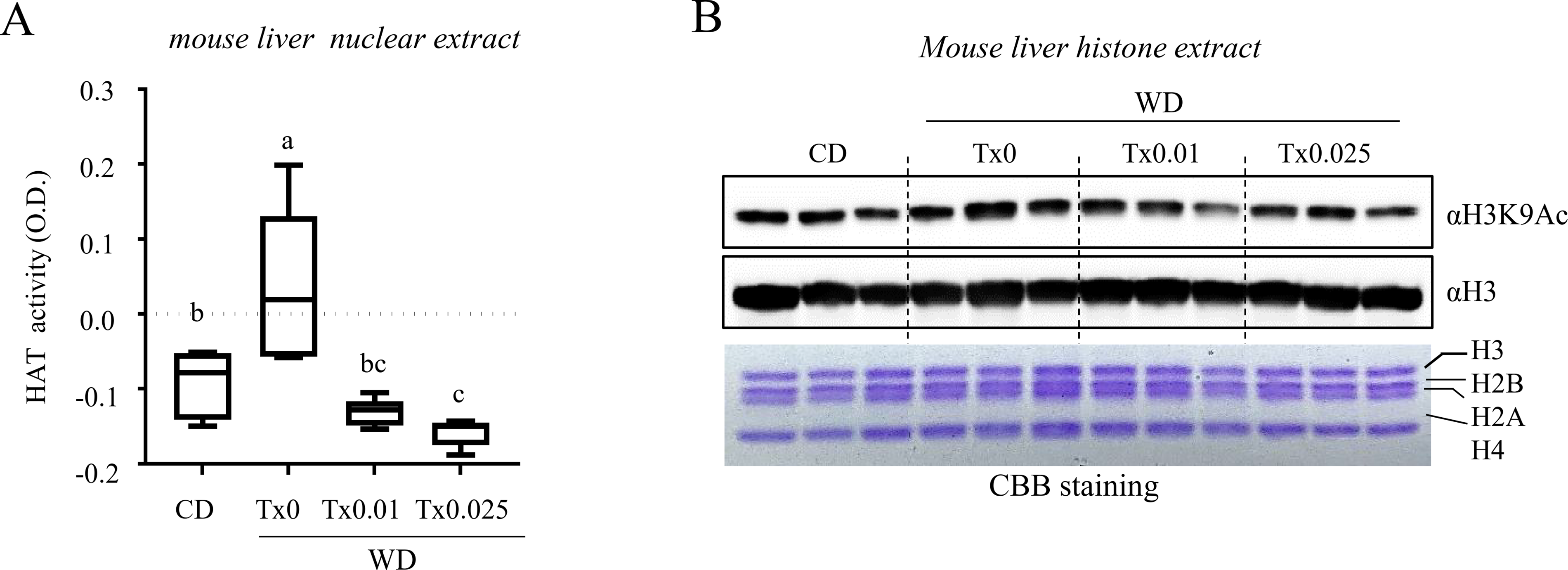
Effects of tamarixetin on histone acetyltransferase (HAT) activity in vivo.
Tx interacts with the acetyl-CoA binding region of the p300 HAT domain
Based on previous HAT activity assay results, we conducted molecular docking simulations to examine how Tx interacts with p300 and influences its enzymatic activity. Docking analysis revealed a stable interaction between Tx and the p300 HAT domain, with a binding affinity of −8.3 kcal/mol for the most stable pose, followed by additional docking poses with binding affinities of −7.9, −6.6, −6.2, and −5.4 kcal/mol (Fig. 3A, Supplementary Table S4, and Supplementary Fig. S2). Structural analysis predicted that Tx localizes to the acetyl-CoA binding site of p300, which was co-crystalized with Lys-CoA, a bisubstrate inhibitor designed to mimic acetyl-CoA and lysine residues in the p300 HAT domain (Fig. 3B). When Tx was co-localized with Lys-CoA, both compounds interacted with key residues within the p300 HAT domain, including L1398, D1399, S1400, V1401, H1402, F1403, R1410, T1411, Y1414, C1438, P1439, P1440, Y1446, Q1455, K1456, I1457, P1458, L1463, W1466, and Y1467 (Fig. 3C). These results provide a structural basis for the possibility that Tx interferes with p300 HAT activity by interacting with the acetyl-CoA-binding region.
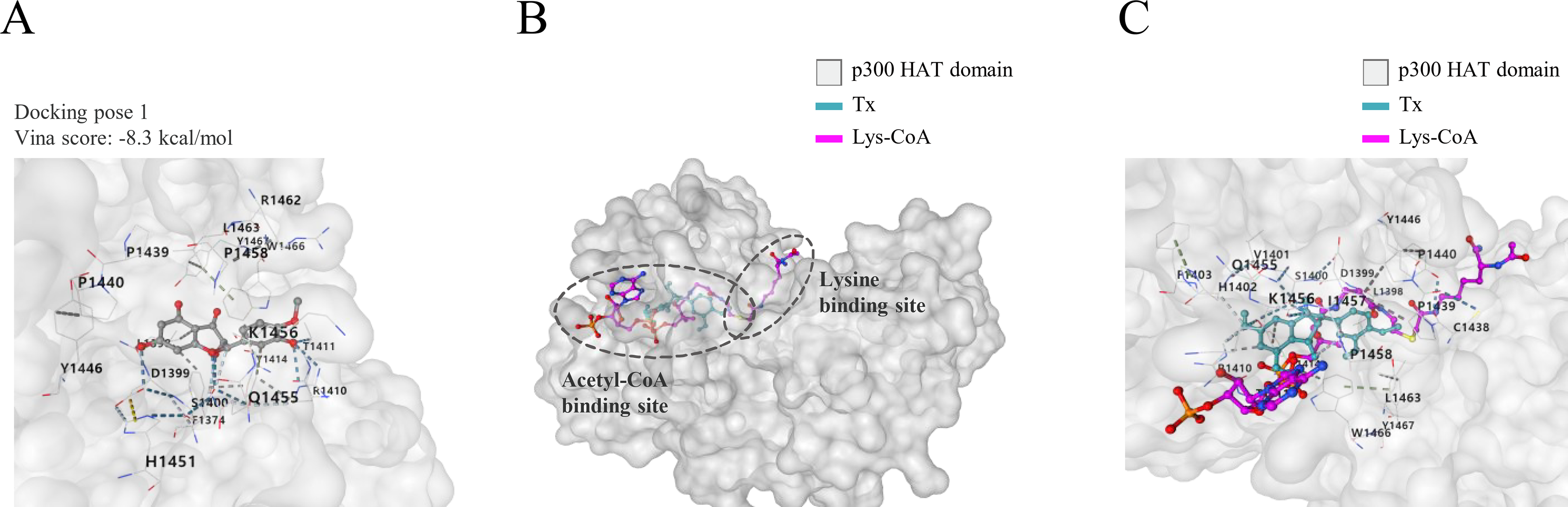
Molecular docking analysis of tamarixetin with the p300 HAT domain.
Tx inhibits H3K9 acetylation by preventing p300 recruitment to the Pdk4 promoter
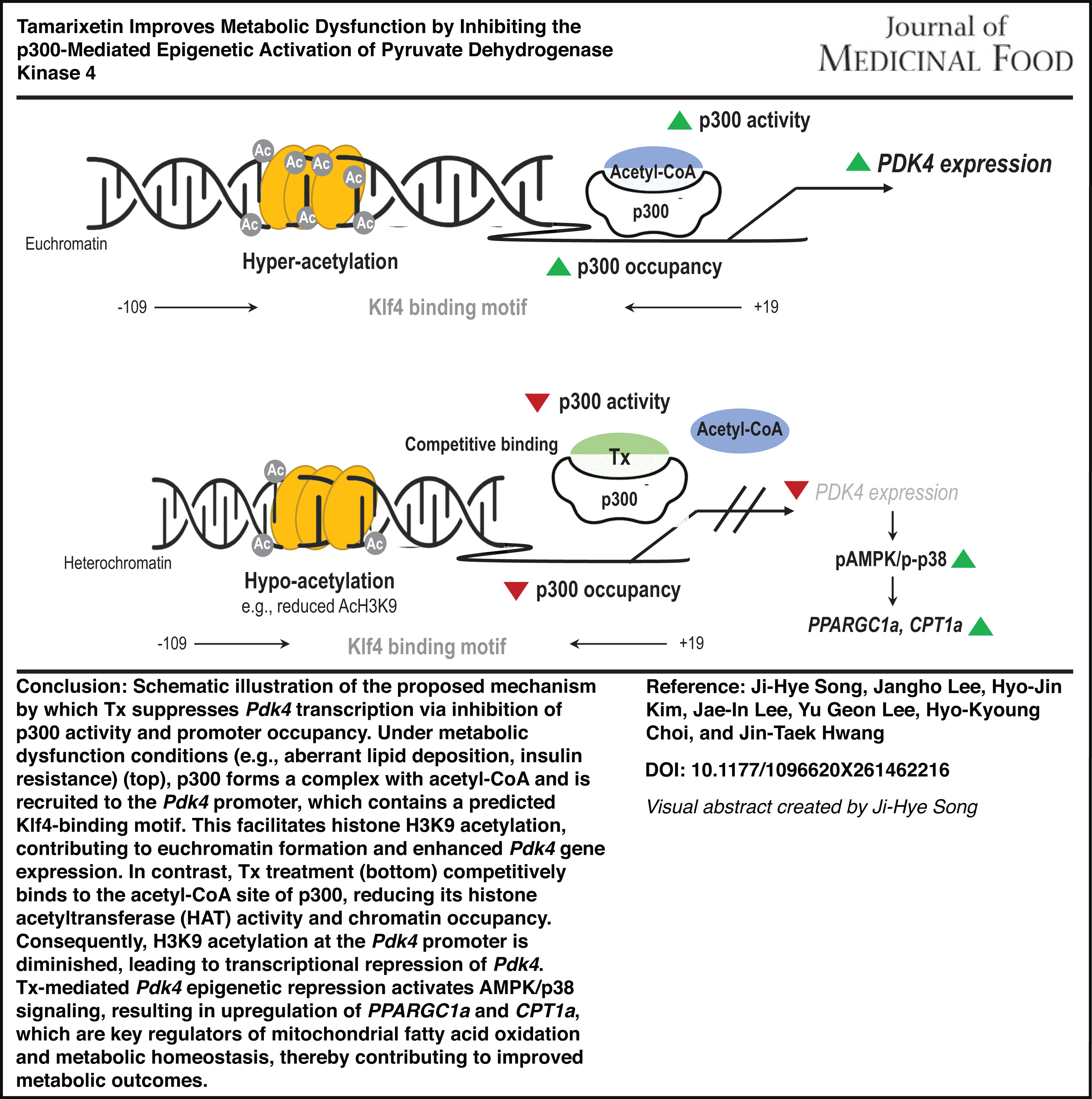
Pdk4 plays a critical role in metabolic regulation, and its transcriptional activation is influenced by epigenetic mechanisms. Based on existing literature and our previous findings, we hypothesized that Tx modulates Pdk4 expression by influencing p300 activity. To investigate this possibility, we performed ChIP analysis to assess p300 occupancy at the Pdk4 promoter. Prior to this, transcription factor (TF)-binding motif enrichment analysis was conducted on the promoter regions (up to −400 base pairs [bp]) of the DEGs to identify potential p300 recruitment sites (Supplementary Table S5). The analysis revealed that Krüppel-like factors (Klf4, Klf5, Klf7, and Klf14) were upregulated in the promoter DEGs following WD supplementation but were subsequently downregulated by WD + Tx treatment. Because p300 has been implicated in the initial stages of adipogenesis through its interaction with Klf4, we examined whether p300 recruitment was altered at Klf4-binding motifs within the Pdk4 promoter. Further analysis of the Pdk4 promoter region revealed a highly conserved Klf4-binding motif within the −400 bp region (Fig. 4A). We then examined changes in p300 occupancy within the region (−109 ∼ +19 bp) containing this motif using ChIP analysis (Fig. 4B, upper panel). As shown in Figure 4B, p300 recruitment increased following WD feeding but decreased after WD + Tx administration. Additionally, histone H3K9 acetylation levels mirrored p300 occupancy within these regions. Overall, these results suggest that Tx suppresses p300 recruitment to Klf4-binding motifs within the Pdk4 promoter, leading to reduced histone H3K9 acetylation in this region. Together, these findings support a locus-specific epigenetic regulatory mechanism linking Tx treatment to the suppression of Pdk4 transcription.
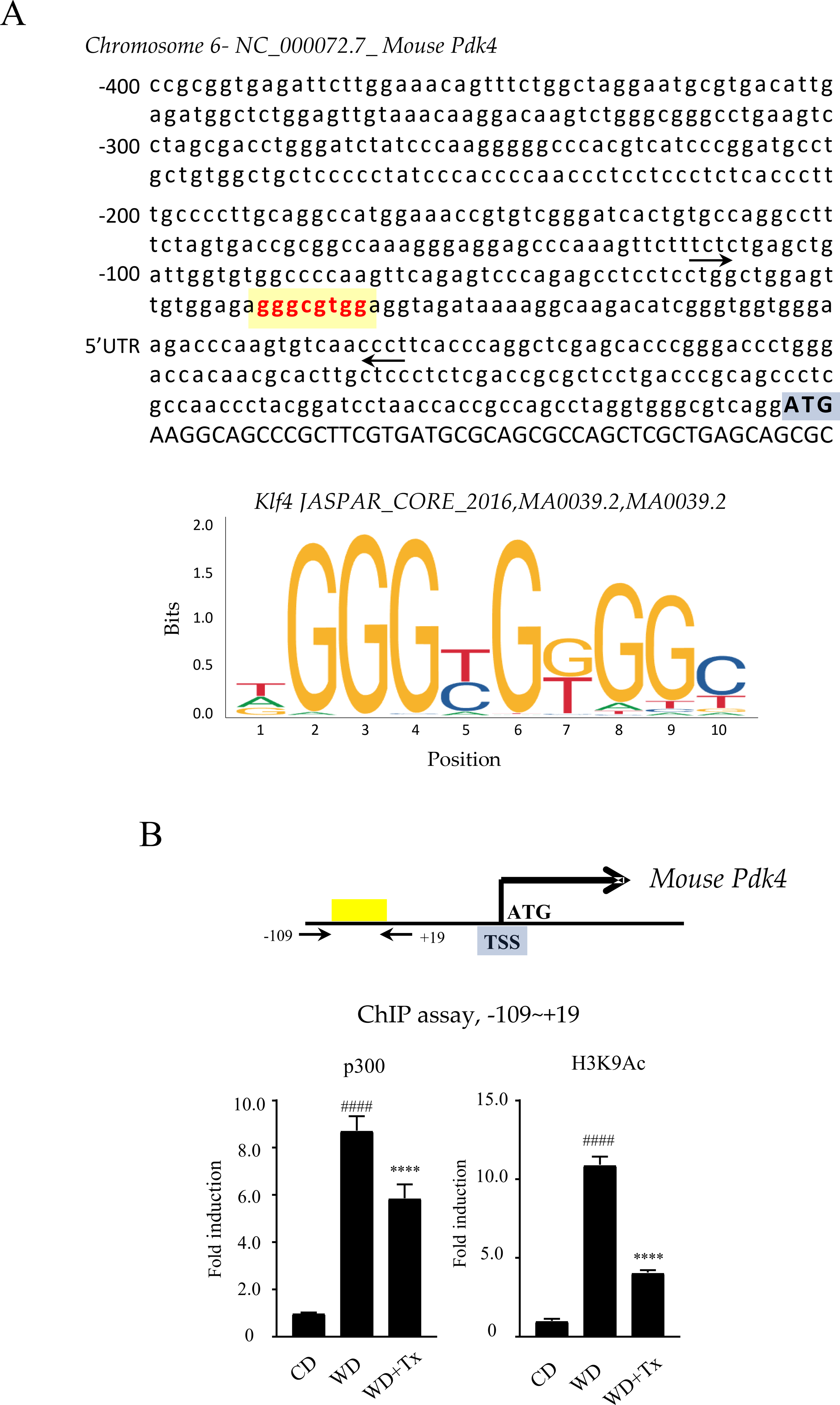
ChIP analysis of p300 and H3K9Ac in the Pdk4 promoter region in vivo
Tx ameliorates WD-induced metabolic dysfunction and insulin resistance in vivo
Next, we examined the effects of Tx supplementation on WD-induced metabolic dysfunction, particularly body weight changes, lipid accumulation, and insulin resistance. Mice in the WD + Tx group exhibited significantly lower weight gain compared to the WD group, with a statistically significant difference emerging during the sixth week of treatment (Fig. 5A). Tx supplementation inhibited weight gain in a dose-dependent manner, with the WD + 0.025% Tx-fed group gaining ∼83% of the weight observed in the WD-fed group (Fig. 5B). Further analysis revealed that Tx supplementation reduced liver enlargement, epididymal fat, and retroperitoneal fat levels in WD-fed mice (Fig. 5C). Both the 0.01% and 0.025% Tx-supplemented groups showed a statistically significant reduction in liver mass compared to the WD group, with values comparable to those of the control group. However, no significant difference was observed between the Tx 0.01% and 0.025% groups. Epididymal and retroperitoneal fat masses were also significantly lower in the Tx-supplemented groups than in the WD group, regardless of Tx concentration. Nonetheless, no significant dose-dependent differences were detected between the Tx-treated groups (Fig. 5D). H&E staining revealed that Tx supplementation reduced lipid droplet size in the liver, epididymal fat, and retroperitoneal fat tissues, with the most pronounced differences observed in hepatic tissue compared to adipose tissue (Fig. 5E). However, circulating triglyceride (TG) concentrations did not differ significantly between groups (Fig. 5F). Lipid profile analysis revealed that total cholesterol and low-density lipoprotein cholesterol (LDL-C) concentrations were significantly lower in the 0.025% Tx group, although HDL cholesterol concentrations also decreased simultaneously (Fig. 5F, Supplementary Fig. S3). Oral glucose tolerance test (OGTT) results showed that blood glucose concentrations were lower in the Tx-treated mice at all measured time points following glucose loading (Fig. 5G). Similarly, Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) values demonstrated improved insulin sensitivity in Tx-treated mice (Fig. 5H). The WD-induced increases in serum AST and ALT levels were significantly attenuated by Tx administration (Fig. 5I). Collectively, these results suggest that Tx supplementation ameliorates WD-induced metabolic dysfunctions and insulin resistance in vivo.
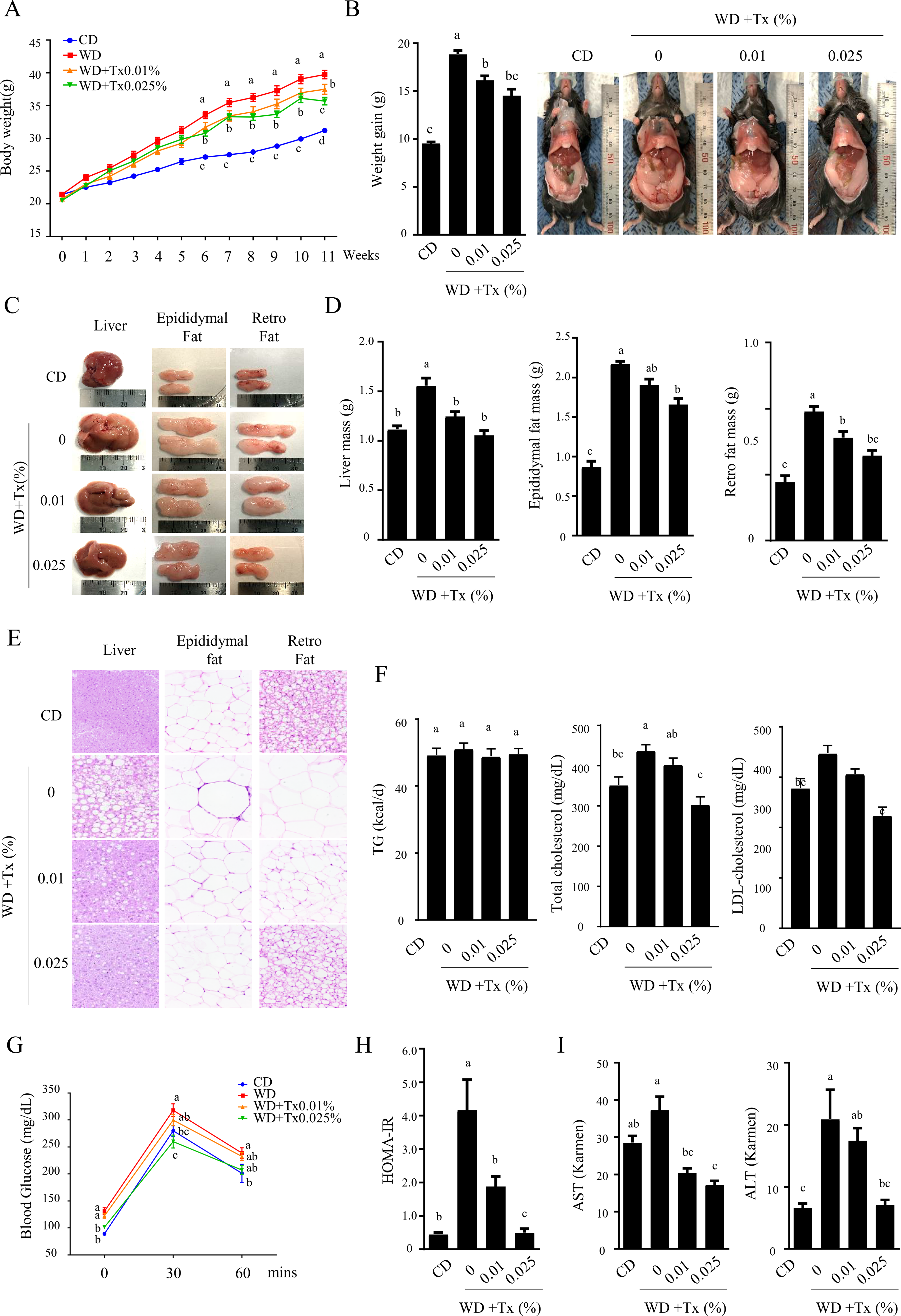
Effects of Tx on Western diet-induced obesity in a mouse model.
Tx-mediated PDK4 suppression is associated with increased expression of insulin-sensitivity-related genes
To determine whether Tx-induced PDK4 suppression influences insulin signaling, we examined the expressions of p38 and AMPK, two key regulators of the insulin signaling pathway. Prior to gene expression analysis, we assessed the cytotoxicity of Tx and OPA in HepG2 cells (Fig. 6A) and evaluated PDK4 expression levels following treatment. Based on the cell viability results, treatment with 6.3, 12.5, and 25 μM of Tx in combination with OPA significantly reduced OPA-induced PDK4 mRNA expression in a dose-dependent manner (Fig. 6B). Next, we assessed the expression of PPARGC1A and CPT1A, genes associated with insulin sensitivity in the liver. As shown in Figure 6C, the phosphorylation levels of p38 and AMPK were elevated following Tx treatment. To further confirm the effects of Tx on insulin sensitivity-related gene expression, we examined the mRNA levels of PPARGC1A and CPT1A. Both genes were upregulated in a dose-dependent manner following OPA + Tx treatment compared with OPA treatment alone (Fig. 6D). A similar expression pattern was observed in cells treated with DCA, a selective PDK4 inhibitor (Fig. 6E). Overall, these findings suggest that Tx-mediated suppression of PDK4 is associated with increased phosphorylation of p38 and AMPK, accompanied by enhanced expression of PPARGC1A and CPT1A, which are linked to improved insulin sensitivity and metabolic regulation.
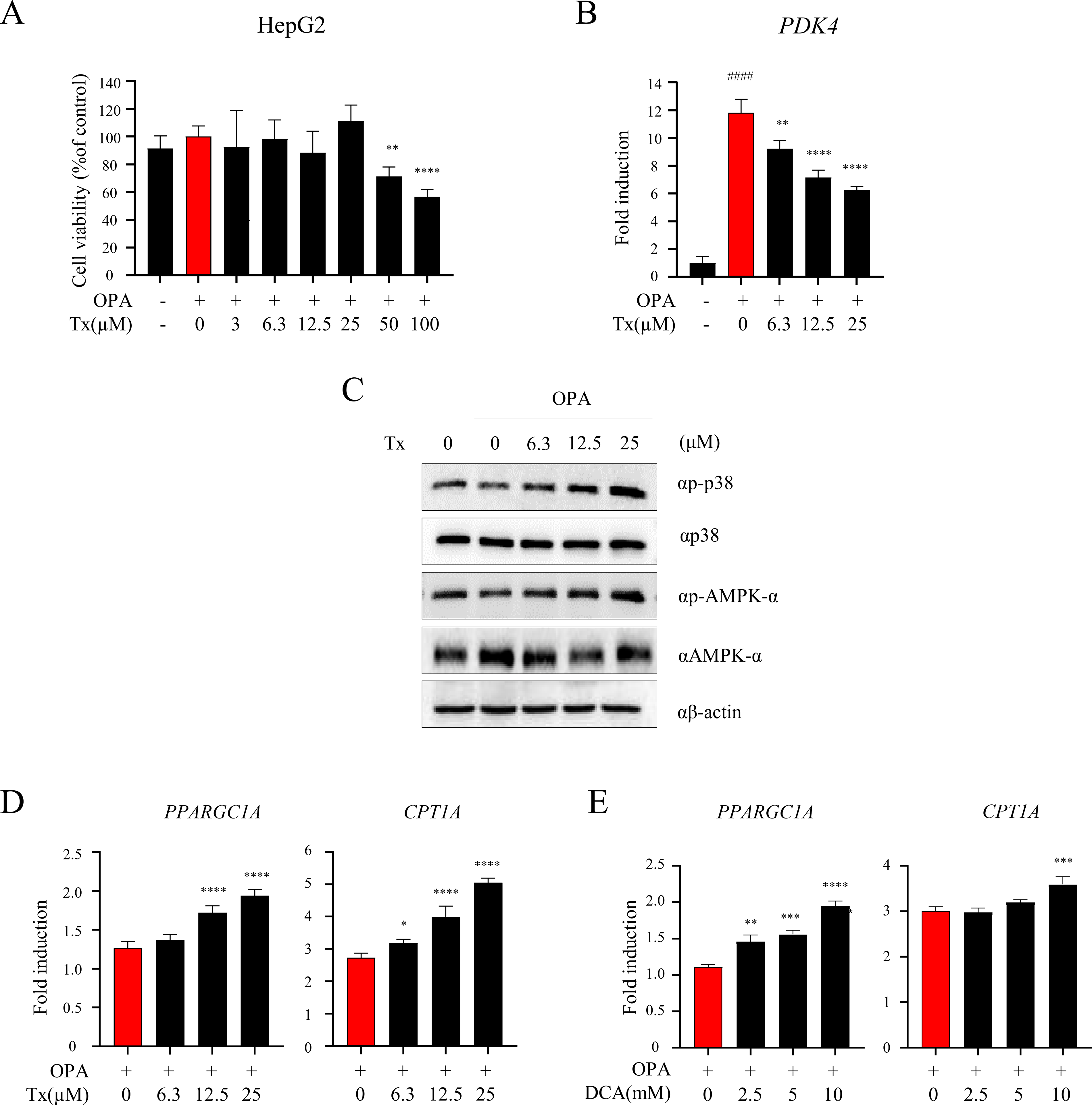
Effects of Tx on PDK4 inhibition, p38 and AMPK signaling, and the expression of target genes PPARGC1A and CPT1A in vitro.
DISCUSSION
In this study, we demonstrated that Tx, a natural flavonoid, ameliorates WD-induced metabolic dysfunction through epigenetic regulation of Pdk4 expression. Tx supplementation effectively reversed hepatic steatosis, adipocyte hypertrophy, hepatomegaly, and insulin resistance despite minimal changes in systemic TG levels. Mechanistically, Tx inhibited the HAT activity of p300 by interfering with acetyl-CoA binding, leading to reduced histone H3K9 acetylation and decreased p300 occupancy at the Pdk4 promoter. This epigenetic repression of Pdk4 was accompanied by activation of the p38/AMPK signaling cascade and upregulation of downstream metabolic genes, such as Ppargc1a and Cpt1a, suggesting that Tx exerts metabolic benefits by linking chromatin-level regulation to lipid metabolism and insulin sensitivity.
Transcriptomic profiling of hepatic tissues from WD– and WD+ Tx-fed mice revealed 10 genes significantly altered by Tx, displaying directional reversals compared to WD. Among them, Pdk4 was selected for mechanistic investigation based on the magnitude of its expression change and its established metabolic role. Pdk4 encodes pyruvate dehydrogenase kinase 4, which inhibits the pyruvate dehydrogenase complex (PDC), thereby suppressing glucose oxidation and promoting lipid utilization. Under metabolic stress, Pdk4 is often upregulated and contributes to insulin resistance, obesity, and hepatic steatosis.8,34–36 Genetic or pharmacologic inhibition of Pdk4 enhances mitochondrial function and lipid clearance, supporting its relevance as a therapeutic target. Histone acetylation is a crucial regulatory mechanism in metabolic disorders.16,17,37 Flavonoids such as Tx can influence chromatin states, particularly histone acetylation, through modulation of HAT or HDAC activity,38,39 and Pdk4 expression is known to respond to histone acetylation at its promoter.20,40,41 Based on these reports, we hypothesized that Tx exerts part of its metabolic benefits through epigenetic suppression of Pdk4. Our findings support this hypothesis, showing that Tx inhibits hepatic HAT activity, reduces H3K9 acetylation, and modulates the transcriptional activity of metabolism-related genes. Although our data demonstrate reduced p300 occupancy and H3K9 acetylation at the Pdk4 promoter, the current study primarily focused on locus-specific epigenetic regulation. Given that p300 broadly regulates histone acetylation at metabolically relevant genes, it remains possible that Tx influences additional transcriptional programs beyond Pdk4. However, genome-wide chromatin occupancy or histone acetylation profiling were not performed in the present study. Therefore, further studies using global epigenomic approaches will be required to define the broader transcriptional landscape regulated by Tx.
p300 is a transcriptional coactivator with intrinsic HAT activity that targets H3K9 and H3K27 residues and functions as a nutrient sensor localized to promoter and enhancer regions of metabolically relevant genes.18,19,42 Pharmacological inhibition of p300 has been shown to mitigate hepatic lipid accumulation, inflammation, and fibrosis in metabolic disease models, indicating that p300 activity responds to nutritional stress.42–44 Our study identified p300 as a candidate HAT regulating H3K9 acetylation at the Pdk4 promoter, particularly because p300 is a major HAT associated with transcriptional activation of metabolic genes through H3K9 acetylation. 42 Consistent with this model, Tx suppressed Pdk4 transcription by reducing H3K9 acetylation and inhibiting p300 occupancy. The computational analysis revealed a high-probability Klf4-binding motif within the Pdk4 promoter overlapping the p300-binding site, suggesting that Tx interferes with a p300-Klf4 complex involved in Pdk4 transcription.45,46 Furthermore, docking analysis revealed that Tx occupies the acetyl-CoA-binding region of the p300 HAT domain, which may contribute to inhibition of its catalytic activity. Inhibition of p300 resulted in reduced histone acetylation and chromatin condensation, leading to decreased recruitment of transcription factors and p300 itself to the Pdk4 promoter. Similar findings were reported for selective HAT inhibition with A-485, which decreased p300 chromatin occupancy, 42 and other studies have shown that p300 occupancy depends on its catalytic activity. Together, these results support a mechanism whereby Tx represses Pdk4 expression through suppression of p300 HAT activity, reduced H3K9 acetylation, and attenuated transcriptional activation.
Pdk4 upregulation contributes to mitochondrial dysfunction and insulin resistance in obesity and diabetes.8,34–36,47 Elevated PDK4 suppresses PDC activity and exacerbates insulin resistance,8,34 whereas its inhibition restores glucose oxidation and metabolic flexibility. In WD-fed mice, pan-PDK inhibition activated hepatic AMPK signaling, enhanced Fgf21 transcription, and reduced obesity and atherosclerosis, while PDK4 knockout alleviated hepatic steatosis through AMPK-dependent suppression of ChREBP.34,35,48 In agreement with these observations, Tx-mediated repression of Pdk4 increased phosphorylation of AMPK and p38 MAPK in the liver. Although direct mediation experiments were not performed in the present study, previous studies have reported that suppression of PDK activity is associated with enhanced AMPK signaling and improved metabolic homeostasis.34–36,48 Consistent with these findings, both Tx treatment and pharmacological inhibition of PDK4 using DCA increased AMPK phosphorylation in our experimental system, supporting a functional relationship between PDK4 suppression and AMPK/p38 activation. Since PDK suppresses PDC activity and limits mitochondrial glucose oxidation, its inhibition enhances oxidative phosphorylation and relieves cellular energy stress, thereby activating AMPK signaling.8,34,35 Activated AMPK and p38 subsequently induce Ppargc1a (PGC-1α), a key regulator of mitochondrial biogenesis and fatty acid oxidation. 49 The upregulation of Ppargc1a and Cpt1a following Tx treatment supports this axis, and increased Cpt1a expression promotes β-oxidation by facilitating long-chain fatty acid transport into mitochondria. These effects may underlie the observed improvements in hepatic steatosis and insulin sensitivity, consistent with reports that AMPK/PGC-1α activation improves metabolic flexibility.8,49
Although the precise mechanisms underlying Tx’s lipid-lowering effects remain incompletely defined, its structural similarity to quercetin provides an important clue. 29 Tx is a monomethoxyflavone methylated at the O-4′ position of quercetin, and quercetin has been reported to improve metabolic disorders by reducing oxidative stress and inflammation and by modulating lipid metabolism. 50 Quercetin downregulates Pdk4, reduces PDC inactivation, enhances pyruvate-to-acetyl-CoA conversion, and increases fatty acid oxidation and energy production, leading to improved insulin sensitivity and reduced lipid accumulation.8,51,52 Given these similarities, Tx may act through a mechanism analogous to quercetin, potentially targeting transcriptional regulation of Pdk4 and related metabolic genes.
Tx, a methylated derivative of quercetin, has been reported to exhibit enhanced metabolic stability and cellular permeability compared with nonmethylated flavonoids. 29 Previous studies have demonstrated that methylated flavonoids possess improved bioavailability and resistance to metabolic degradation, supporting their therapeutic potential in metabolic disease contexts.29,30
Despite the lack of significant changes in plasma TG levels, WD-fed mice displayed classical features of metabolic dysfunction, including increased body weight, hepatomegaly, adipocyte hypertrophy, hepatic lipid accumulation, and insulin resistance. This disconnect suggests that systemic TG concentrations may not fully capture lipid dysregulation, as early metabolic derangements appear primarily in tissues before being reflected in circulating biomarkers.1,2 Moreover, increased intracellular lipid content rather than circulating TG has been shown to correlate more strongly with insulin resistance.3,4 Consistent with this, WD-fed mice exhibited significant increases in serum total cholesterol and LDL-C, whereas Tx supplementation, particularly at 0.025%, significantly reduced both parameters. These findings suggest that Tx selectively modulates cholesterol metabolism through mechanisms distinct from TG regulation. Taken together, these results establish Tx as a promising dietary flavonoid with therapeutic potential against metabolic dysfunction. Through the suppression of Pdk4 transcription via p300 inhibition and downstream activation of AMPK/p38-PGC-1α signaling, Tx improves insulin sensitivity and reduces hepatic lipid accumulation. Although additional work is needed to clarify pharmacokinetic properties, upstream signaling pathways, and comparative efficacy with existing PDK4 inhibitors, this study provides the first evidence supporting the involvement of p300-mediated histone acetylation in Tx-mediated repression of Pdk4. These findings not only provide mechanistic insight into the metabolic benefits of Tx but also lay the groundwork for its development as a novel therapeutic candidate for obesity-associated metabolic disorders.
AUTHORS’ CONTRIBUTIONS
Conceptualization: H.-K.C. and J.-T.H. Data curation: J.-T.H. Funding acquisition: H.-K.C. and J.-T.H. Investigation: J.-H.S., H.-J.K., and J.L. Methodology: J.L., J.-H.S., J.-I.L., and Y.G.L. Project administration: H.-K.C. and J.-T.H. Supervision: H.-K.C. and J.-T.H. Writing of the original draft: H.-K.C. Validation: H.-K.C. Writing, review, and editing: H.-K.C. and J.-T.H.
Supplemental Material
sj-docx-1-mef-10.1177_1096620X261462216 — Supplemental material for Tamarixetin Improves Metabolic Dysfunction by Inhibiting the p300-Mediated Epigenetic Activation of Pyruvate Dehydrogenase Kinase 4
Supplemental material, sj-docx-1-mef-10.1177_1096620X261462216 for Tamarixetin Improves Metabolic Dysfunction by Inhibiting the p300-Mediated Epigenetic Activation of Pyruvate Dehydrogenase Kinase 4 by Ji-Hye Song, Jangho Lee, Hyo-Jin Kim, Jae-In Lee, Yu Geon Lee, Hyo-Kyoung Choi, and Jin-Taek Hwang
Footnotes
DATA AVAILABILITY
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
AUTHOR DISCLOSURE STATEMENT
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by the Main Research Program (
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.