
Abstract
Background:
Preeclampsia contributes significantly to pregnancy-associated morbidity and mortality as well as future risk of cardiovascular disease in mother and offspring, and preeclampsia in offspring. The lack of reliable methods for early detection limits the opportunities for prevention, diagnosis, and timely treatment.
Purpose:
The purpose of this study was to explore distinct DNA methylation patterns associated with preeclampsia in both maternal cells and fetal-derived tissue that represent potential biomarkers to predict future preeclampsia and inheritance in children.
Method:
A convenience sample of nulliparous women (N = 55) in the first trimester of pregnancy was recruited for this prospective study. Genome-wide DNA methylation was quantified in first-trimester maternal peripheral white blood cells and placental chorionic tissue from normotensive women and those with preeclampsia (n = 6/group).
Results:
Late-onset preeclampsia developed in 12.7% of women. Significant differences in DNA methylation were identified in 207 individual linked cytosine and guanine (CpG) sites in maternal white blood cells collected in the first trimester (132 sites with gain and 75 sites with loss of methylation), which were common to approximately 75% of the differentially methylated CpG sites identified in chorionic tissue of fetal origin.
Conclusion:
This study is the first to identify maternal epigenetic targets and common targets in fetal-derived tissue that represent putative biomarkers for early detection and heritable risk of preeclampsia. Findings may pave the way for diagnosis of preeclampsia prior to its clinical presentation and acute damaging effects, and the potential for prevention of the detrimental long-term sequelae.
Preeclampsia, a syndrome unique to pregnancy, is characterized by new-onset hypertension and significant proteinuria in the second half of pregnancy (Fabry, Richart, Chengz, Van Bortel, & Staessen, 2010; Roberts, Pearson, Cutler, & Lindheimer, 2003). Preeclampsia affects approximately 1–8% of pregnancies (Hutcheon, Lisonkova, & Joseph, 2011; Roberts et al., 2003), the prevalence having increased by more than 50% over the last two decades ( Martin et al., 2011). Higher rates of preeclampsia are reported among women who are nulliparous, at extremes of maternal age or have a multiple gestation. In addition, cardiovascular risk factors, such as race and obesity, are associated with risk of preeclampsia (Lo, Mission, & Caughey, 2013). Representing the third leading cause of pregnancy-associated morbidity and mortality worldwide (Ghulmiyyah & Sibai, 2012), preeclampsia is associated with systemic vascular dysfunction and poor placental perfusion presenting later in pregnancy. However, the pathologic state of preeclampsia is thought to begin in the early weeks of gestation as a subclinical condition involving abnormal placentation, representing the etiology of later endothelial dysfunction, altered coagulation, and a heightened inflammatory state. Early prevention, diagnosis, and treatment of preeclampsia are limited by the absence of reliable biomarkers to detect preeclampsia prior to manifestation of classic clinical symptoms (Conde-Agudelo, Villar, & Lindheimer, 2004; Gruslin & Lemyre, 2011). The development of reliable, accurate screening methods and tools for early diagnosis has the potential to improve clinical outcomes through identification of mechanistic underpinnings that would reveal novel treatment targets.
The impact of preeclampsia goes beyond the immediate pregnancy outcomes. A history of preeclampsia represents increased risk of developing cardiovascular disease later in life in both women who survive preeclampsia (Barton & Sibai, 2008; Berends et al., 2008; Carty, Delles, & Dominiczak, 2010; Harskamp & Zeeman, 2007) and their offspring (Anderson, 2007; Geelhoed et al., 2010; Lampinen, Ronnback, Kaaja, & Groop, 2006; Mangos, 2006; Marin et al., 2000; Ray, Vermeulen, Schull, & Redelmeier, 2005; Roberts & Hubel, 2010; Smith, Pell, & Walsh, 2001; Staff, Dechend, & Pijnenborg, 2010; Wilson et al., 2003). Additionally, daughters of women with preeclampsia have a 2-fold increased risk of developing preeclampsia during their pregnancies (Skjaerven et al., 2005). Sons born to mothers with preeclampsia are also more likely to father a child who is a product of a preeclamptic pregnancy (Esplin et al., 2001), providing evidence of a genetic component to heritable preeclampsia risk. To date, no single candidate gene has been identified that offers clinical utility for identification of those at risk of preeclampsia or future sequelae. Due to our limited mechanistic understanding of the syndrome, the absence of identified susceptibility genes (Chappell & Morgan, 2006), and heritable risk that follows a complex, non-Mendelian transgenerational inheritance pattern (Williams & Broughton Pipkin, 2011), identification of epigenomic biomarkers for preeclampsia may be of prognostic value and could generate detailed information facilitating the rational design of new therapeutics. Therefore, it is imperative to investigate the underpinnings of preeclampsia that may represent both the cause of disease and the heritability of risk for this heterogeneous condition.
Epigenetic mechanisms of gene regulation are heritable and influenced by environmental factors. DNA methylation of the CpG nucleotide bases is a major epigenetic event that can influence the regulation of gene expression in the development, differentiation, and aging and is responsible for the maintenance of specific, heritable patterns of gene expression in humans (Jones & Baylin, 2007; Klose & Bird, 2006; Weber, Stresemann, Brueckner, & Lyko, 2007). These CpG sites may be located within the gene promoter, gene body, or flanking island, shelf or shore regions (Irizarry, Wu, & Feinberg, 2009). Although most studies of DNA methylation have focused on the promoter regions, recent evidence suggests that the altered DNA methylation in the CpG island shore regions is strongly related to gene expression.
Most DNA methylation patterns are established in utero, though cells may continue to remodel chromatin and establish new expression patterns during postnatal development (Lahiri, Maloney, & Zawia, 2009; Loke et al., 2013; Zawia, Lahiri, & Cardozo-Pelaez, 2009). Methylation patterns established during fetal life may be affected by placental insufficiency, which limits the utilization and availability of key nutrients including those impacting methylation (e.g., folate, methionine, and cysteine). Recent reports suggest that global DNA methylation is increased in placental tissue samples of patients with preeclampsia (Jia et al., 2012; Kulkarni, Chavan-Gautam, Mehendale, Yadav, & Joshi, 2011), and hypomethylation in promoter regions of multiple genes may be an indicator and/or consequence of early-onset preeclampsia (Yuen, Penaherrera, von Dadelszen, McFadden, & Robinson, 2010). To date, no published reports of differential DNA methylation in preeclampsia linking mother and offspring are available, limiting evidence to correlate the fetal epigenome with the maternal intrauterine environment.
In addition to the impact on short-term gene expression, epidemiological evidence suggests that environmental exposures during development may play a role in disease susceptibility later in life. Researchers have hypothesized that epigenetic changes in gene regulation are responsible for this phenomenon (Herceg, 2007; Jirtle & Skinner, 2007). As DNA methylation patterns are largely established during fetal life, epigenomic alterations represent a plausible explanation of the heritable development of preeclampsia and cardiovascular disease resulting from placental insufficiency. Therefore, the purpose of this study was to identify distinct DNA methylation patterns in women during early pregnancy that may predict preeclampsia and in fetal-derived placental tissue that may represent heritable changes in the epigenome established during fetal life.
Method
Participant Recruitment
In this prospective study, we recruited a convenience sample of nulliparous women in the first trimester of pregnancy from a local community. Signage in community locations where pregnant women were likely to be (e.g., health care agencies, grocery stores, and fitness centers) was supplemented by advertisement in print and televised media to communicate the opportunity for study participation. Eligibility criteria included no previous deliveries, age > 18 years, and pregnancy at less than 14 weeks’ gestation. Research team members screened interested candidates, provided study details to eligible women, and enrolled them upon receiving their consent. The Institutional Review Boards of the University of North Dakota and the cooperating health system approved the study, and human subjects’ protection was assured throughout the study.
Sample and Data Collection
We collected maternal peripheral blood (MP-B) from participants via venipuncture in the first trimester of pregnancy. White blood cells were isolated, providing the source for the DNA used to quantify methylation in this study. Upon delivery, placental tissue was placed in cold physiological saline solution. After removal of decidua, chorionic tissue was collected from 6 individual sites equally spaced along the placental periphery in morphologically normal areas and stored at −80°C until analyses. Samples obtained from the 12:00 position were homogenized for analyses.
Pregnancy outcome was determined by medical record abstraction. Preeclampsia was identified by documented diagnosis or evidence of new-onset hypertension (systolic blood pressure [BP] ≥ 140 mmHg or diastolic BP ≥ 90 mmHg) combined with proteinuria (≥ + 1 single sample or > 300 mg/24-hr urine sample) during the second half of pregnancy or during the postpartum hospitalization (Roberts et al., 2003). Achievement of diagnostic criteria in relation to gestational age determined subcategorization of early- versus late-onset preeclampsia (
Sample Analyses
DNA was purified from maternal peripheral blood and placental tissue with phenol-chloroform-isoamyl alcohol (Life Technologies Corporation) using established isolation procedures (Ohm et al., 2010). Approximately 3 μg of total genomic DNA was sent to the Illumina-certified Biomedical Genomics Core at the University of Minnesota for Illumina Infinium DNA methylation 450K bead-based array analysis (Bibikova et al., 2011; Dedeurwaerder et al., 2011; Sandoval et al., 2011). Genome-wide DNA methylation data were normalized, and differentially methylated CpG sites were identified using the GenomeStudio DNA methylation module (Illumina). Average beta scores for each individual CpG site were compiled for normotensive control and preeclampsia samples (n = 6/group). The Infinium platform uses a bead-based array to identify the percentage of methylation at any single CpG dinucleotide locus. Average beta scores were assigned based on the percentage of methylation at individual loci within a sample and ranged from 0 (0% methylation) to 1 (100% methylation) within a given sample. Sites without measureable beta scores across any of the 12 samples were discarded. Individual CpG dinucleotides having a change in beta (delta.beta) score in the preeclampsia group compared to the normotensive control group of greater than 0.2 (indicating > 20% increase in methylation compared to controls) and a p value < .05 by two-tailed t-test were defined as having significantly increased methylation. CpG dinucleotides with a delta.beta across preeclampsia samples of <−0.2 (indicating >20% decrease in methylation compared to normotensive controls) and a p value < .05 were defined as having significantly decreased methylation. Samples were also independently analyzed using the NIMBL (Numerical Identification of Methylation Biomarker Lists). Infinium analysis package for Matlab provided by Frank Wessely, School of Veterinary Medicine and Science, University of Nottingham, UK. The NIMBL package is publically available and requires Matlab (tested using Matlab release version 7.11) and the Statistics and Bioinformatics Matlab toolboxes. NIMBL has been specifically designed to identify biomarkers in clinical samples by identifying spots with maximum absolute distance between control and experimental samples, taking into account the heterogeneity that is common in clinical samples (Wessely & Emes, 2012). NIMBL and NIMBL-qc were performed on all maternal white blood samples. The top 20 differentially methylated sites were identified, with a maximum of two preeclampsia samples masked to allow for heterogeneity.
Results
Participant Demographics
A total of 64 women were enrolled, with 86% (N = 55) retained through the duration of the study. Reasons for not completing the study included moving from the area (n = 2), miscarriage (n = 2), changed to nonparticipating provider (n = 1), and lost to follow-up (n = 4). Of the women who completed the study, 12.7% (n = 7) developed preeclampsia. First-trimester maternal white blood cell (average gestational age of 64.5 days across all samples) and placenta samples were available for six of the women with preeclampsia and were included in the DNA methylation analysis, matched with samples from six women with normotensive pregnancy outcomes based upon maternal age (range 0–9 years difference) and weight at first prenatal visit (range 1–20 lb difference).
Demographic data describing study participants included in DNA methylation analyses are shown in Table 1. Gestational weight gain was significantly greater among women who developed preeclampsia (p = .038), though there were no significant group differences in other weight measures in mothers or infants. BP did not differ significantly between the two groups across three trimesters of pregnancy; however, there was a significant increase in systolic and diastolic BP on postpartum Day 1 (Figure 1) in the women with preeclampsia compared to those with a normotensive pregnancy. The timing of hypertension onset, coupled with an absence of significant differences in pregnancy duration and infant birth weight, indicates the more common syndrome of late-onset preeclampsia.
Sample Characteristics.
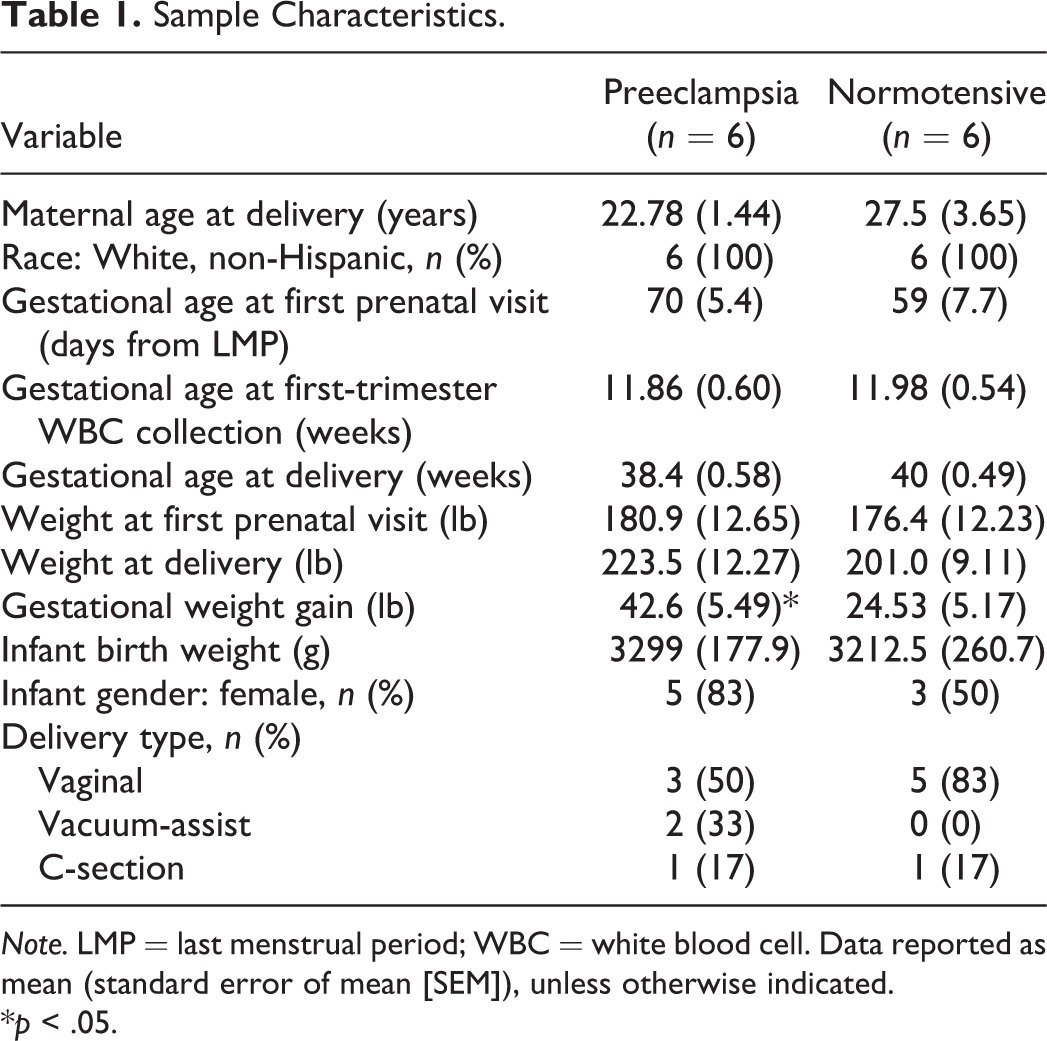
Note. LMP = last menstrual period; WBC = white blood cell. Data reported as mean (standard error of mean [SEM]), unless otherwise indicated.
*p < .05.
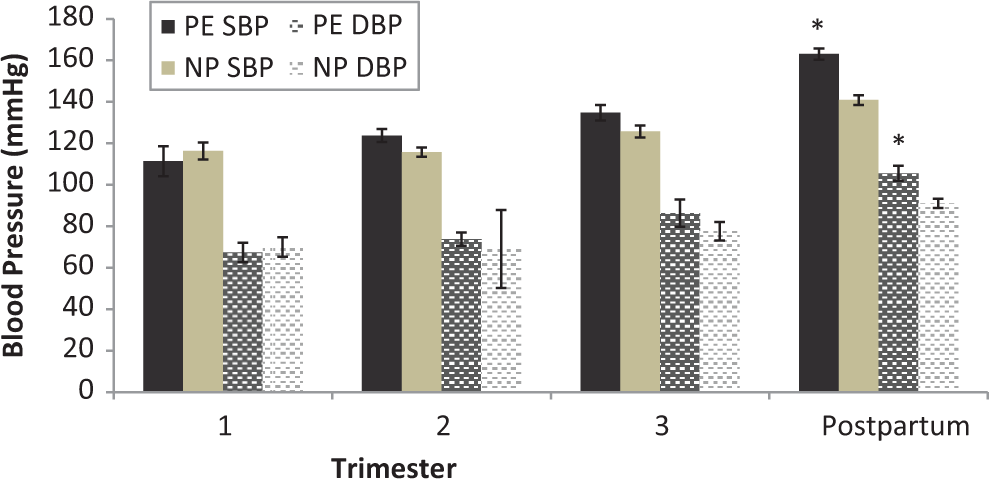
Maternal blood pressure. Systolic blood pressure (SBP) and diastolic blood pressure (DBP) during pregnancy did not differ significantly between women with preeclampsia (PE) and those with normotensive pregnancy (NP). SBP and DBP were both significantly higher in women with preeclampsia compared to women with normotensive pregnancy in the early postpartum period. *p < .05 compared to normotensive group.
Identification of Potential DNA Methylation Biomarkers in Maternal Peripheral Blood
A total of 207 CpG dinucleotides were identified as being differentially methylated in the maternal peripheral blood cells in women who developed preeclampsia as compared to those who were normotensive (Figure 2A), including both gain and loss of methylation at individual CpG dinucleotides. Of these identified sites, 64% showed a gain of methylation in preeclampsia (top cluster, Figure 2B), while 36% were associated with a loss of methylation (bottom cluster, Figure 2B). For sites either gaining or losing methylation, these CpG dinucleotides were distributed across most chromosomes in the genome (Figure 2B, left panels). The majority of these differentially methylated sites were not associated with known CpG islands (Figure 2B, middle panels) and were found to be either within the body of known genes or in areas of the genome that are not currently known to be associated with any annotated gene (Figure 2B, right panels). The remainder of differentially methylated CpG sites was found in CpG islands or flanking shore and shelf regions (Doi et al., 2009; Figure 2B, middle panels). Biological pathways in genes differentially methylated in preeclampsia were determined by cluster analysis (Table 2). Genes with significant gain in methylation were associated with cell signal transduction involving lipid binding, protease enzyme inhibition, protein–protein interaction, cell cycle processes, and adhesion. Associations with signaling pathways involving cellular metabolic processes predominated in genes that had significant methylation loss.
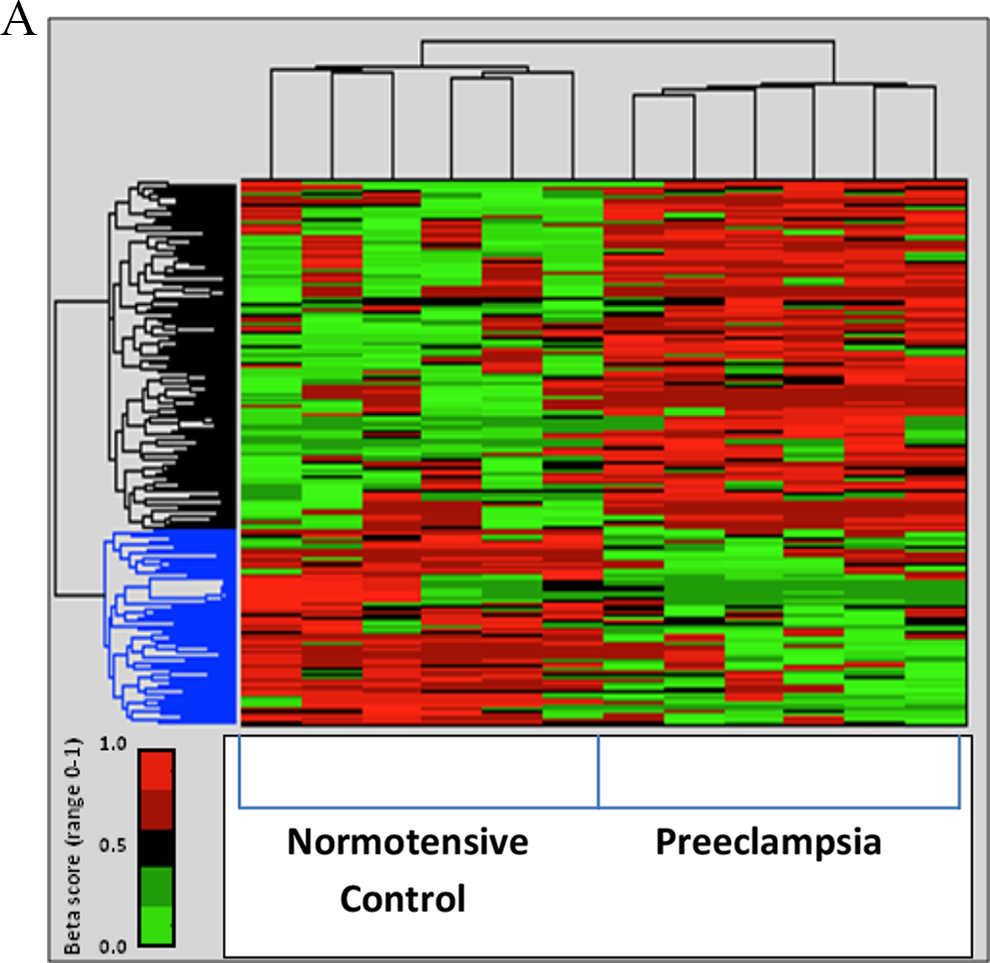
Both gain and loss of methylation at individual CpG dinucleotides can be observed in the first trimester in the peripheral blood of women who will develop preeclampsia during pregnancy. A. Heatmap showing differential methylation of CpG sites in maternal peripheral blood using the Illumina Infinium 450K methylation arrays. A total of 207 individual CpG dinucleotides are shown that were identified as gaining methylation (associated with a change in average beta score [delta.beta] > 0.2; top cluster—black) or losing methylation (delta.beta < −0.2; bottom cluster—blue) in women with preeclampsia (PE) as compared to normotensive controls (C). (p value of < .05 in maternal peripheral white blood [M-PB] cells.) B. Characterization of identified CpG dinucleotides in the gain and loss of methylation clusters from maternal peripheral blood including chromosomal distribution, relation to known CpG islands using University of California, Santa Cruz (UCSC) designation, and distribution of sites in relation to known genes according to UCSC_Refgene_Group association. For each, total number of either CpG sites or known genes is shown.
Biological Pathways Associated With Differentially Methylated Genes in Preeclampsia.

Note. First-trimester maternal white blood cell and placenta samples for six of the women with preeclampsia were matched with samples from six women with normotensive pregnancy outcomes and were included in the DNA methylation analysis. Of the 133 CpG dinucleotides with gain of methylation, 71 known genes were identified. Applying high stringency, the functional cluster with the highest enrichment score included genes involved in pleckstrin homology, small domains that occur in a large variety of signaling proteins, where they serve as simple targeting domains that bind lipids. Methylation loss in 74 CpG dinucleotides was associated with 38 known genes. Signaling pathways involved endothelin signaling, T-cell activation, insulin signaling, progesterone-related oocyte maturation, MS-related immune injury, phospholipase C-epsilon, G-protein signaling (Gi, Gs), encephalin release, and metabotropic glutamate receptor group 1 pathway in genes with significant methylation loss. Associated diseases included type II diabetes, Huntington disease, and cystic fibrosis.
To further narrow the focus to DNA methylation biomarkers with the greatest potential for clinical utility and to test whether this analysis can successfully identify CpG sites with potential clinical biomarker utility, we performed additional Infinium methylation array analyses using the NIMBL software package written using Matlab (Wessely & Emes, 2012). NIMBL compares differentially methylated sites and corresponding genes between two groups of samples. The NIMBL data analysis tool also contains a module (NIMBL-qc) that allows for a quality assessment of samples. Briefly, multiple output plots are generated to visualize the sample quality. This analysis includes visualization of beta value distribution of each sample and measures deviation from the expected distribution, largely related to the detection of p values. The identification of low-quality samples is performed using a Kolmogorov–Smirnov test. All of the samples used in this study passed NIMBL-qc, as did unmethylated and fully methylated control samples (data not shown). Differential methylation of top array sites is shown in Figure 3. We identified 20 CpG sites as putative biomarkers for early screening of preeclampsia, of which 13 were associated with known genes (Table 3). Of the 13 known genes that we identified with NIMBL analysis, 10 met the initial delta.beta criteria and 12 had significant differences in mean methylation in maternal white blood cells.
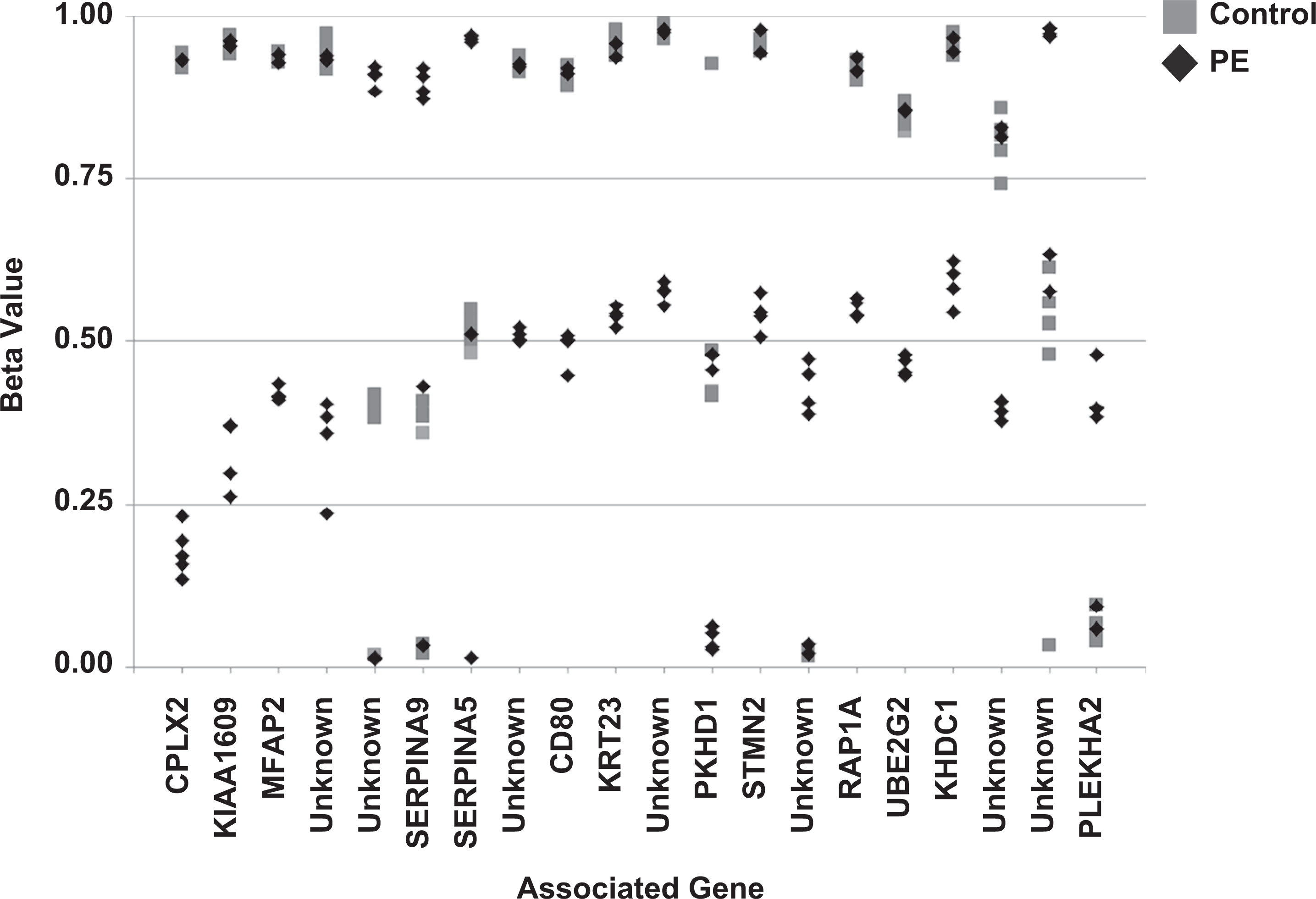
Identification of top 20 sites for differential methylation of maternal peripheral white blood cells as potential DNA methylation biomarkers for early detection of preeclampsia (PE) in women. Sites were identified using Infinium’s Numerical Identification of Methylation Biomarker Lists (NIMBL) software package based on a minimum absolute difference in beta scores between the normotensive control group and the preeclampsia group, adjusting for the masking of up to two preeclampsia samples to allow for heterogeneity in clinical samples.
Potential Early Screening Biomarkers in Preeclampsia.
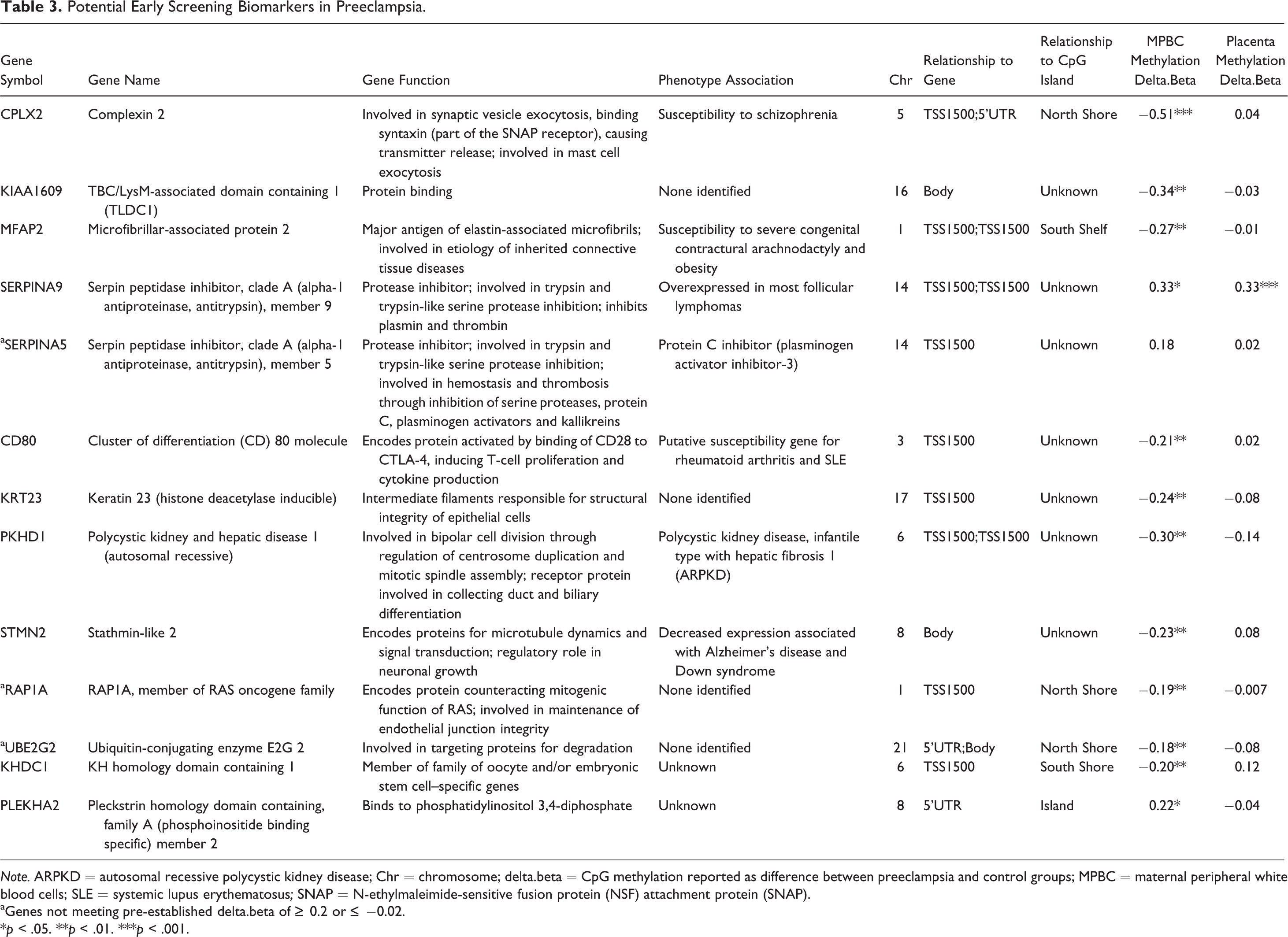
Note. ARPKD = autosomal recessive polycystic kidney disease; Chr = chromosome; delta.beta = CpG methylation reported as difference between preeclampsia and control groups; MPBC = maternal peripheral white blood cells; SLE = systemic lupus erythematosus; SNAP = N-ethylmaleimide-sensitive fusion protein (NSF) attachment protein (SNAP).
aGenes not meeting pre-established delta.beta of ≥ 0.2 or
*p < .05. **p < .01. ***p < .001.
Interpretation of DNA methylation data and evaluation of potential biomarkers is complex due to the heterogeneity of primary clinical samples. The NIMBL platform permits masking of a limited number of samples to allow for biomarker identification despite heterogeneity among samples. Figure 3 illustrates such heterogeneity, where one or two preeclampsia samples may cluster with normotensive controls for each spot tested, despite the statistically significant differences in average beta score between the two groups and the clear separation of the majority of preeclampsia samples from control in terms of potentially informative sites. These data underscore the need to perform this type of secondary analysis and not simply rely on average beta scores, as it is clear from this analysis that all individual patients are not expected to have 100% of the abnormal biomarkers despite statistically significant differences in beta scores between the two groups. We expect that the most successful and clinically evaluable DNA methylation biomarker panels will need to include multiple DNA methylation sites and will rely on clinically evaluable distance in average beta that distinguishes preeclampsia from normotensive pregnancy.
Potential Transmission of DNA Methylation Biomarkers of Preeclampsia From Mother to Offspring During Pregnancy
Finally, we wanted to test whether DNA methylation across the genome may play a role in facilitating the transgenerational transmission of preeclampsia risk from parent to offspring. In order to begin to assess whether DNA methylation changes are also seen in offspring of women with preeclampsia, we isolated DNA from the placenta at the time of delivery and measured global methylation changes using the Illumina Infinium platform as described previously. CpG dinucleotides identified as being differentially methylated in MPBCs (Figure 2) were analyzed for methylation changes in placental tissue from women with preeclampsia as compared to normotensive controls (Figure 4). The majority of the 64% of differentially methylated sites in MPBCs that were associated with a significant gain of methylation also showed varying amounts of methylation gain in placenta tissue, with many sites showing significant methylation gains in placenta based on a delta.beta > 0.2 (Figure 4; shades of red, left placental panel). Further, the 36% of differentially methylated sites in MPBCs that were associated with a significant loss of methylation were likely to also show a loss of methylation to varying degrees in placental tissue (shades of green; right placental panel), with many sites showing significant methylation losses based on a delta.beta ≤−0.2 (Figure 4; right panel).
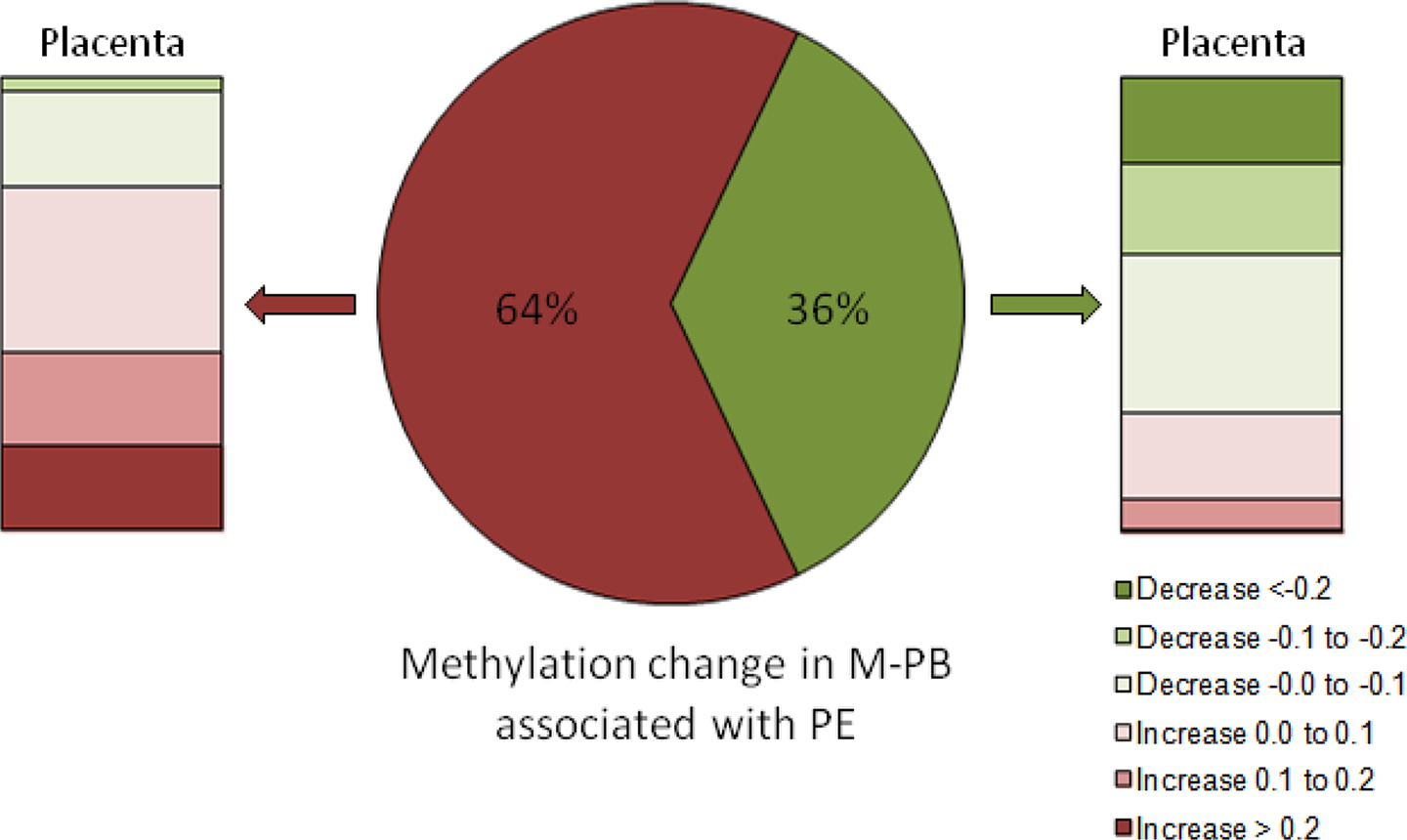
Potential transmission of DNA methylation biomarkers of preeclampsia (PE) from mother to child during pregnancy. Of the differentially methylated sites in maternal peripheral white blood cells (M-PB), 64% were associated with a significant gain of methylation (red, center pie chart), and the majority of these sites also showed varying amounts of methylation gain in placental tissue (shades of red; left panel). The 36% of the differentially methylated sites in M-PB that were associated with a significant loss of methylation (green, center pie chart) were, similarly, more likely to show a loss of methylation to varying degrees in placental tissue (shades of green; right placental panel).
Discussion
Preeclampsia cannot currently be diagnosed until later in pregnancy when women manifest the characteristic signs and symptoms. Clinical indications used to diagnose preeclampsia include BP exceeding 140 mmHg systolic or 90 mmHg diastolic with significant proteinuria of >300 mg/24 hr. However, by the time these indications are present, significant systemic injury has already been established. Researchers have not yet identified a biologically sound mechanistic explanation of how preeclampsia develops, a gap in knowledge that hinders the development of a reliable method for early screening and diagnosis in the clinical setting. Methods currently employed to diagnose preeclampsia include sonographic placental maturation (Walker, Hindmarsh, Geary, & Kingdom, 2010), ultrasound uterine artery Doppler pulsatility indices (Plasencia et al., 2011; Stampalija, Gyte, & Alfirevic, 2010), and serum markers (Martin & Brown, 2010; Wortelboer et al., 2010) along with identification of clinical indicators. A recently developed method that measures angiogenic factors in maternal serum (PlGF, placental growth factor; sFlt-1, soluble fms-like tyrosine kinase-1) modeled significant cost savings over the traditional BP and proteinuria diagnostic criteria, combining Doppler and serum uric acid measurements (Hadker et al., 2010). However, low sensitivity and specificity of this method limits the clinical utility.
The purpose of the present study was to identify evidence for differential DNA methylation in pregnancies complicated by preeclampsia. To that end, we quantified genome-wide DNA methylation in individual CpG sites, identifying significant differences in methylation in maternal white blood cells collected during the first trimester of pregnancy and in placental tissue between women who subsequently developed preeclampsia and those with normotensive pregnancies. Our findings demonstrate, for the first time, differential methylation of novel genes in maternal white blood cells and placental tissue in preeclampsia. These findings may represent DNA methylation biomarkers for preeclampsia. Development of a selective and specific biomarker panel would assist in identifying individuals at increased risk of developing preeclampsia who may benefit from early intervention strategies and may also lead to the development of effective interventions. Further, screening prior to pregnancy may be used to predict future risk of preeclampsia-complicated pregnancy.
The NIMBL method we employed may prove useful for the development of a DNA methylation biomarker panel, with either increased or decreased methylation in preeclampsia, that can be used in the future for detection of preeclampsia using maternal peripheral blood as early as the first trimester. The specific putative biomarkers we identified with the NIMBL method have not been previously reported in the preeclampsia literature (see Table 3). However, several of the genes do play an important role in pregnancy, miscarriage, implantation, or immune tolerance. Stathmin family and KH homology domain genes are important in pregnancy and implantation (Schulz, Widmaier, Qiu, & Roberts, 2009; Tian, Pascal, Fouchecourt, Pontarotti, & Monget, 2009). The E2 class of ubiquitin-conjugating enzyme is upregulated in early miscarriage (Liu et al., 2006). CD80 is vital for maternal immune tolerance to the fetus (Abumaree, Chamley, Badri, & El-Muzaini, 2012; Moldenhauer, Keenihan, Hayball, & Robertson, 2010). Finally, Liu et al. (2012) noted that RAP1A protein was downregulated in gestational diabetes mellitus. Additionally, researchers have reported that other members of the gene families of some of these putative biomarkers are differentially expressed in preeclampsia. For example, genes in the SERPIN family, and in particular SERPINA3, have been implicated in preeclampsia (Blumenstein et al., 2009). However, there are no reports of differential expression or regulation of SERPINA5 or SERPINA9 in preeclampsia.
Though we have focused on differential methylation in maternal peripheral blood here due to the potential to use this tissue for early detection, our analysis of DNA methylation in fetal-derived placental tissue also clearly revealed differential methylation when comparing tissue from normotensive pregnancies and that from pregnancies complicated by preeclampsia, suggesting that DNA methylation changes may be propagated across generations. Other investigators have identified differential methylation in single genes in either placental tissue or maternal sera, though none, to our knowledge, have reported finding differentially methylated genes common to both mothers and fetal tissue. Müller et al. (2004) reported a significantly higher percentage of differential methylation in the APC gene from cell-free DNA in sera of women during the first trimester who subsequently developed preeclampsia compared to women who did not develop the condition. As both maternal and fetal cell-free DNA have been reported in maternal sera, the authors could not definitely determine the source of the DNA (maternal versus fetal). The APC gene is involved in pathways that counter metastasis. Increased methylation in early pregnancy among women who developed preeclampsia may be associated with the impaired trophoblast invasion that occurs during placental development in preeclampsia, leading to placental insufficiency. Our novel finding of common differences in methylation in maternal blood taken in the first trimester of pregnancy and in fetal-derived placental tissue has the potential to change clinical practice, improving health outcomes for mothers and their children.
DNA methylation is associated with changes in gene expression, particularly when the differentially methylated CpG dinucleotides are associated with gene promoter regions (e.g., CpG islands), marking the start of gene transcription. In the present study, we identified differential methylation in areas that flank the promoter regions, including shores and shelves. Research in other disease states has shown that these regions are also potentially linked to gene expression changes (Doi et al., 2009). Additional work is needed to understand the significance of CpG sites located outside of CpG islands but within the promoter regions of known genes (5′UTR, TSS1500, TSS200, first exon) and in gene bodies or regions of the genome not known to be associated with any annotated genes in terms of gene expression changes. Studies conducted in animal models have clearly shown that early-age environmental stimuli can affect epigenomic patterning and lead to gene expression changes that increase disease risk in adulthood (Lillycrop, Phillips, Jackson, Hanson, & Burdge, 2005; Weaver, Diorio, Seckl, Szyf, & Meaney, 2004). The methylation changes we reported here may still be heritably transmitted from mother to offspring and may serve as clinical DNA methylation biomarkers of preeclampsia regardless of gene expression changes.
Our laboratory previously reported evidence of vascular dysfunction and hypertension across two generations of male and female offspring in a rat model of placental insufficiency (Anderson, Lopez, Zhang, Pavlish, & Benoit, 2005a, 2005b; Anderson, Lopez, Zhang, et al., 2006; Anderson, Lopez, Zimmer, & Benoit, 2006) consistent with heritable patterns of cardiovascular disease after preeclampsia. The induction of epigenetic changes in a cell by environmental stimuli has been implicated in a variety of human diseases associated with aging, including cancer, infertility, and multiple neurodegenerative disorders (Donkena, Young, & Tindall, 2010; Lahiri et al., 2009; Ren et al., 2011; Tunc & Tremellen, 2009; Zawia et al., 2009). Recently, researchers have begun to explore whether these heritable epigenomic changes might also play a role in vascular disorders such as stroke (Qureshi & Mehler, 2010), hypertension (Arngrimsson, 2005; Wang, Prins, Sober, Laan, & Snieder, 2011), and cardiovascular disease (Xiao & Ye, 2011). Lofton Day et al. (2008) explored DNA methylation biomarkers for blood-based screening for colorectal cancer risk, narrowing the list of putative biomarkers to three candidate genes.
The strengths of the present study include the prospective design, which provided evidence of the potential for DNA biomarkers that precede the overt manifestations of preeclampsia. The use of a biomarker panel based on DNA methylation would provide objective evidence for diagnosis of preeclampsia, assuring prompt treatment of women with preeclampsia while avoiding unnecessary treatment of women who do not meet diagnostic criteria. In order for DNA methylation to be used as a specific and selective screening method for preeclampsia, a cost-effective panel of select markers will be required for validation. While the sample size of the present study is a potential limitation, the power to identify significant differences provides evidence of the robust effect, minimizing concerns of power in this exploratory study. Additional studies are ongoing to determine if these DNA methylation biomarkers may also be heritable across multiple generations and predictive of the increased preeclampsia and/or associated cardiovascular disease risk reported in families with individuals who have been diagnosed with preeclampsia. The successful identification of a DNA methylation biomarker for preeclampsia would provide novel opportunities to improve clinical care of families across generations.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The support provided by the Robert Wood Johnson Foundation Nurse Faculty Scholar Award (CMA, #64202).