
Abstract
Background:
Alport syndrome is an inherited Type IV collagenopathy characterised by renal failure, hearing loss and ophthalmic manifestations such as lenticonus and dot-and-fleck retinopathy. New signs have been described which can be useful both for diagnosis and for prognosticating the risk of complications. This study examines and describes a triad comprising the unusual ‘stair-case’ foveal sign, together with choroidal thinning and late-stage peripheral schisis in a patient with Alport syndrome.
Case presentation:
This is a case report of a 49-year-old Caucasian male with a background of X-linked Alport syndrome presenting with gradual and progressive diminution of vision in the left eye with a central blur. He had already undergone three renal allografts, was deaf and suffered from hypertension by the time of his first presentation to ophthalmology. On examination, corrected visual acuity was 6/9.5 in the right eye and 6/30 in the left eye. Optical coherence tomography imaging showed an unusual ‘stair-case’ sign of the fovea in both eyes, together with choroidal thinning. We postulate that an abnormal vitreomacular interface followed by vitreomacular traction and eventually separation, removing layers of the inner retina with the vitreous, led to this unusual appearance. Subsequently, this patient also developed schitic changes more peripherally in the retina which progressed over the following 5 years.
Conclusion:
The stair-case foveal sign, choroidal thinning and mid-peripheral schisis are three signs that clinicians might expect to encounter on optical coherence tomography imaging of patients with Alport syndrome. These findings can be attributed to unique mutations of collagen IV which lead to a variety of clinical phenotypes affecting basement membrane structures. Identification of these features may not only be useful diagnostically and in forecasting complications such as macular holes, but also predict mode of inheritance and likelihood of early-onset renal failure.
Introduction
Alport syndrome is an inherited Type IV collagenopathy characterised by mutations in COL4A3 and COL4A4 genes in the autosomal recessive subtype and COL4A5 in the X-linked subtype. 1 It affects at least 1 individual in every 10,000. Diagnosis is imperative due to the risk of disease in family members, with early medical treatment delaying the onset of end-stage renal failure.
The syndrome is phenotypically heterogeneous, yet is characterised by haematuria, renal failure, loss of hearing and ocular abnormalities which can affect the cornea, lens and the retina. 2
The ocular features in Alport syndrome can be rationalised by the mutations in the collagen IV α3α4α5 network which cause basement membrane thinning, lamellation and rarefaction, alongside the intraocular mechanical stresses. Hence, membranes in the cornea are weak and adherence to the epithelium, endothelium and underlying stroma is poor, leading to problems such as recurrent corneal erosion syndrome 3 and posterior polymorphous corneal dystrophy. 4 The absence of the α3α4α5 network in the lens leads to protrusion of the lens through the weakest and thinnest component of the capsule, that is, lenticonus and small ruptures can also lead to cataract formation after healing occurs. 5
In the retina, the most commonly described abnormality is a peripheral fleck retinopathy, with optical coherence tomography (OCT) appearances suggesting an abnormality between the retinal pigment epithelium (RPE) and Bruch’s membrane. 6 Although visual acuity (VA) is usually normal with this finding, it is associated with early-onset renal failure. 7 Central fleck retinopathy has also been described, which is associated with thinning of the inner limiting membrane (ILM) and disruption of the ILM/nerve fibre layer, which incorporates the α3α4α5 network. Again, VA is usually normal with this finding. Other retinal features described include macular retinoschisis, 8 bull’s eye maculopathy, vitelliform macular detachment and macular holes. 9 The stair-case sign has been previously reported by our group in 2013 and is proposed to arise from rarefaction of the inner retina and superficial choroid. 10
Identifying these ocular signs can help differentiate inheritance and subtype of Alport syndrome. For example, a peripheral fleck retinopathy is more commonly identified in men with X-linked Alport syndrome, and central retinopathy in a female is more attributable to autosomal recessive Alport syndrome. 7
Case description
A Caucasian male aged 49 years was first diagnosed with Alport syndrome at the age of 15 after persistent haematuria and sensorineural deafness. Subsequent renal biopsy in 1975 yielded mild mesangial proliferation, though immunofluorescence was negative. The patient underwent three renal transplants for his renal disease, requiring long-term oral antihypertensives, immunosuppressants and anticoagulants. His systemic hypertension was well controlled henceforth. Genetic testing identified his subtype as being X-linked Alport syndrome.
He presented to ophthalmology with a 6-month history of blurred vision in the left eye. His best-corrected VA was 6/9.5 in the right and 6/30 in the left. He was moderately myopic, with a refraction of R: −4.50/−2.00 × 180; L: −4.50/−2.00 × 180. There was neither lenticonus, nor cataract, although he did suffer from some dry eye symptoms. He had a dull macular reflex on fundoscopy and bilateral maculopathy, which was corroborated by fundus imaging and autofluorescence (Figure 1).
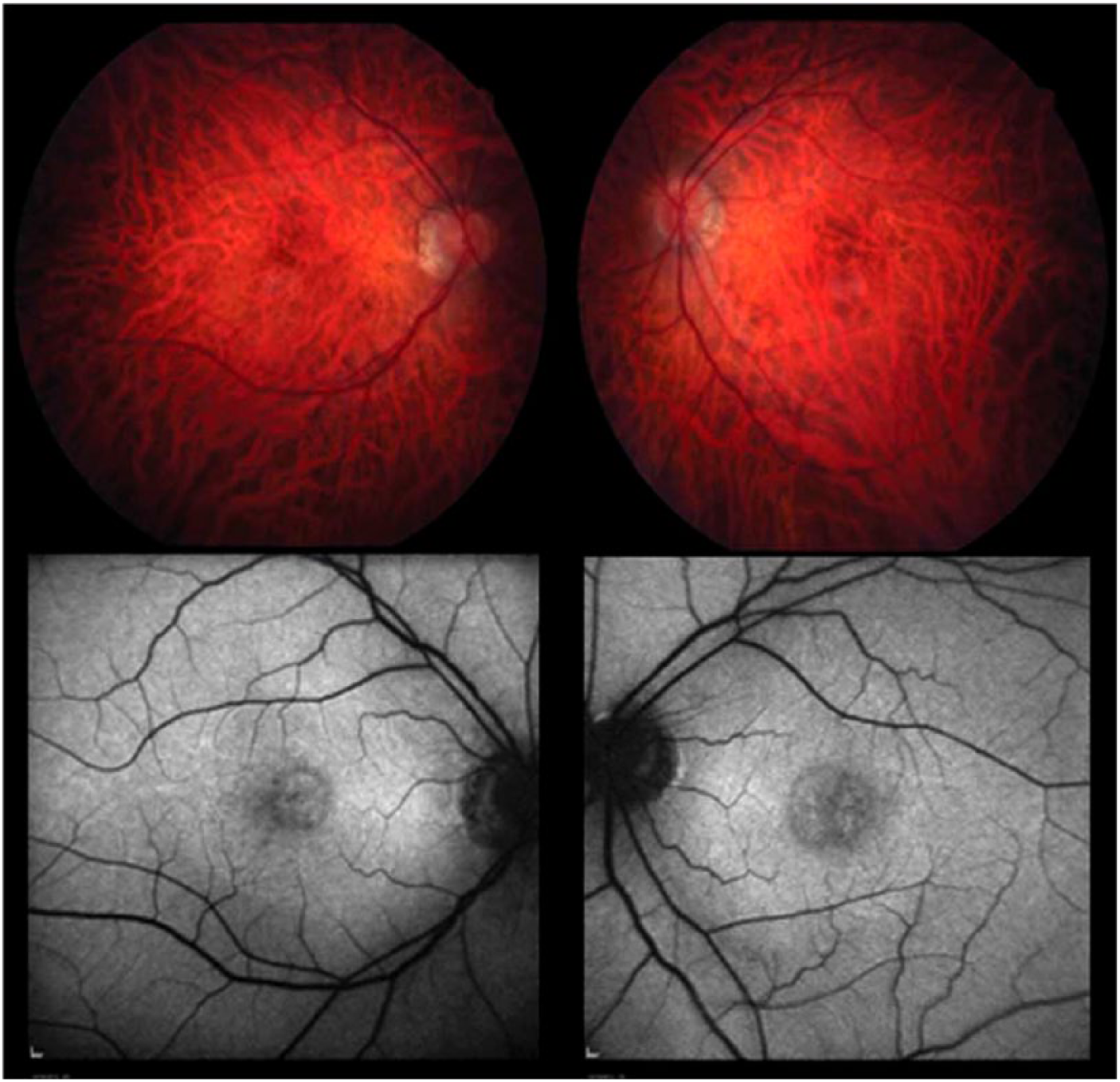
Colour fundus photographs (top) and autofluorescence images (bottom). Colour fundus photos show bilateral maculopathy and dull macular reflexes. Autofluorescence shows disruption of normal pattern of foveal autofluorescence.
OCT imaging yielded an unusual feature characterised by an irregular pattern of foveal thinning, with loss of the usual clivus contour and an irregular foveal edge demonstrating a ‘stair-case’ pattern. The patient was subsequently observed over a period of 4 years, during which time his vision remained stable. However, there is continued progression of the ophthalmic features on OCT (Figure 2).
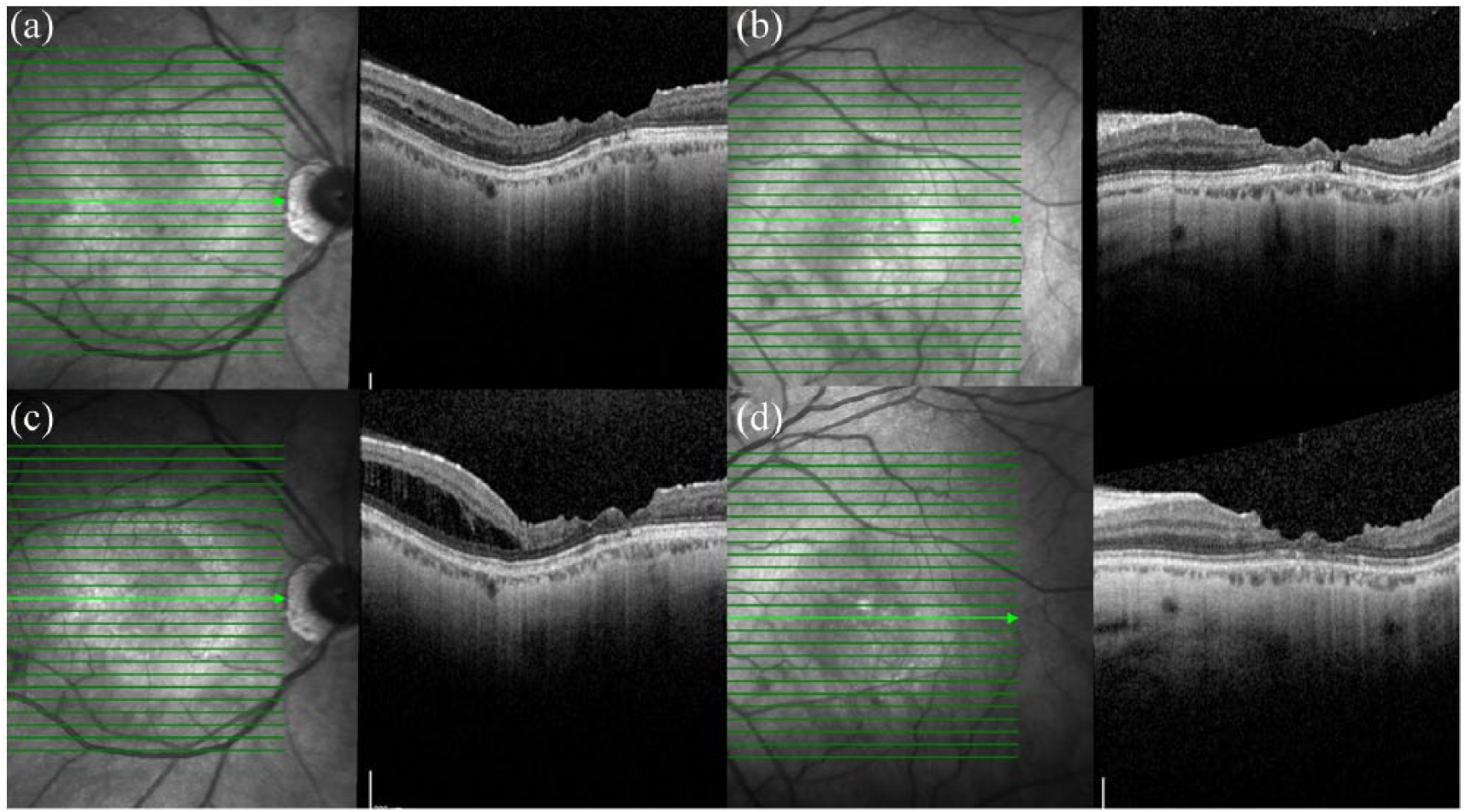
OCT images of the right (a) and left (b) maculae, showing the ‘stair-case’ foveal sign – irregularity of the clivus profile and foveal margin. OCT images of the right (c) and left (d) maculae 5 years after initial presentation, also showing the ‘stair-case’ foveal sign in both eyes. In the right eye (c), there has been a development of schisis in the temporal macula.
Schitic changes in the peripheral macula were also noted (Figure 3).
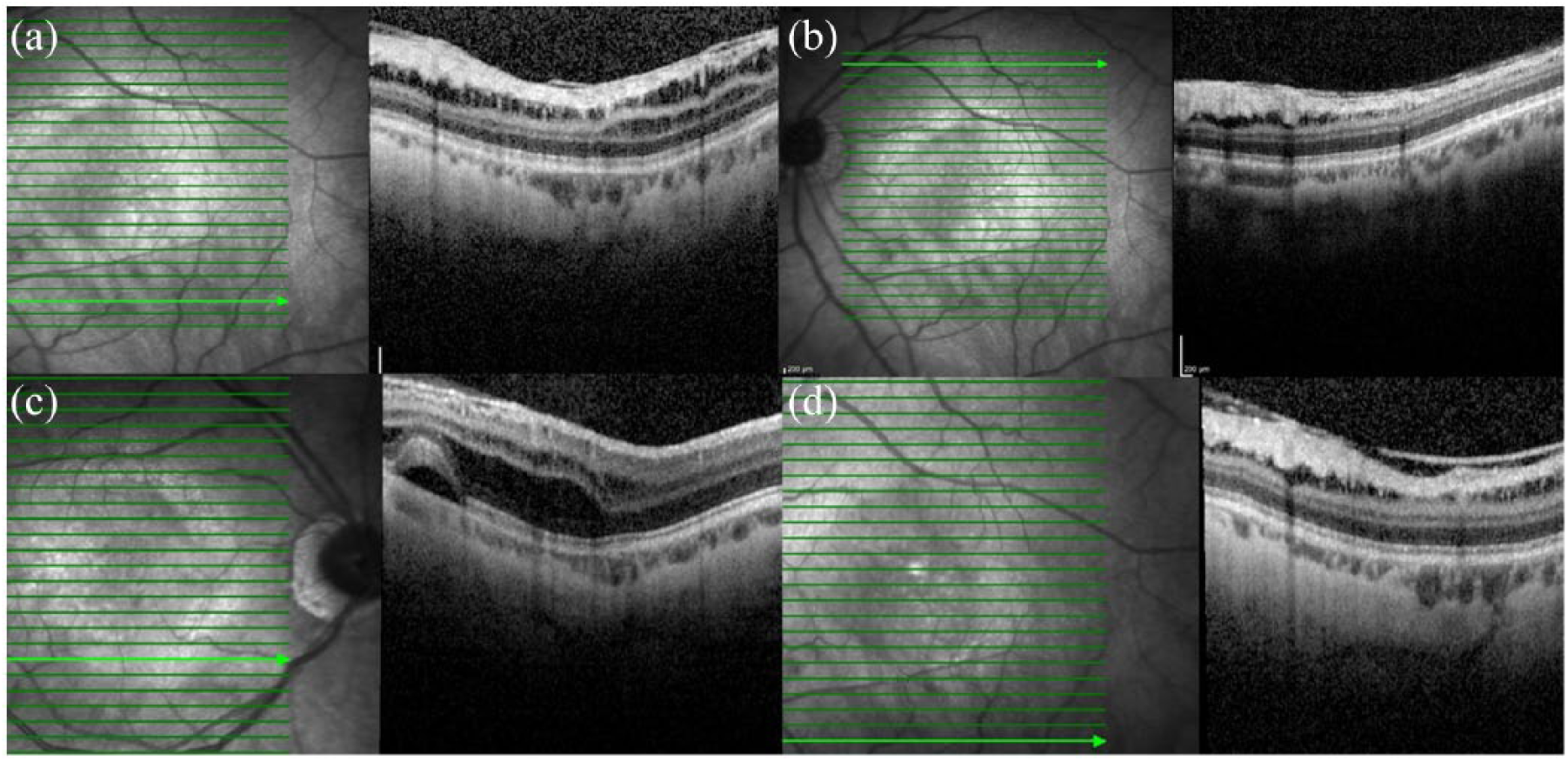
More profound schisis peripherally, within the left macula (a, b). Further schitic changes can be seen slightly more peripherally in the right eye (c), though the left remains largely unchanged (d).
The choroidal thickness was quantitatively measured using an in-built calliper within the spectral domain OCT machine (Spectralis) and was found to be markedly reduced (Figure 4). This showed thinning of the inner choroid, most evident subfoveally and varying between 49 and 100 μm within 2500 μm of the foveal centres, although this may be confounded by the patient’s bilateral moderate myopia.
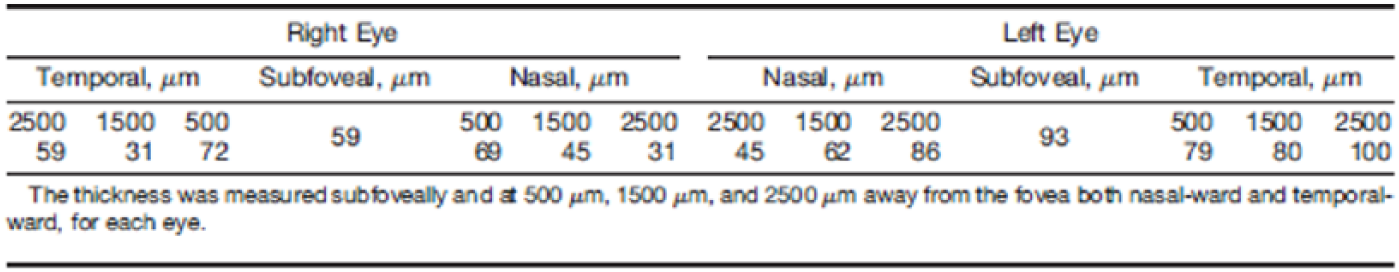
Markedly reduced choroidal thickness, measured with spectral domain OCT machine.
Despite progression in the schitic changes on OCT, both centrally and peripherally, the balance of risk favours observation for this patient (unless the patient develops a complication such as a macular hole, necessitating a vitrectomy). Notably, the patient’s eye discomfort became more severe and he was subsequently diagnosed with recurrent corneal erosion syndrome which required extensive lubrication.
Conclusion
This is the first case reporting this triad of features in a patient with Alport syndrome. The unusual ‘stair-case’ foveal sign, reported previously, 10 is the result of an abnormal vitreomacular interface brought about by an abnormal Type IV Collagen within the ILM and intervening extracellular matrix, which is then followed by vitreomacular traction and subsequent separation, removing layers of both the inner retina and the vitreous. This may be a precursor of the macular holes which are associated with Alport syndrome. In these instances, any decision to intervene with vitrectomy should be taken cautiously, as such a chronic degenerative process may have a bad prognosis following surgery, 11 wherein the lack of a normal retinal scaffold can have a significant impact when removal of the ILM or epiretinal membrane is performed. These procedures can lead to a variety of morphologic changes which may bring about further adverse consequences on retinal architecture and function. 12
This study also quantifies the thinning of the inner choroid, showing significant thinning subfoveally and varying between 49 and 100 μm within 2500 μm of the foveal centres, although this may be confounded by the patient’s bilateral moderate myopia. Choroidal thickness can also be decreased in patients who suffer from systemic hypertension 13 with or without chronic renal disease. However, as the patient’s hypertension was well controlled, and in the absence of any other systemic diseases 14 (such as sickle cell retinopathy), the reduction in choroidal thickness is likely to be due to structural changes caused by Alport syndrome. We propose that such thinning can be analogous to nephropathy caused by thin basement membranes and may therefore predict risk of renal failure.
Retinoschisis has been described in other patients with Alport syndrome; 9 however, this is the first case which reports schitic changes in the periphery rather than the macula. Our case supports the hypothesis that the Type IV collagenopathy in Alport syndrome and impairment of the basement membrane of RPE–Bruch’s membrane complex may lead to passage of the fluid into the retina which results in schitic changes. In addition, the loss of the collagen IV α3α4α5 network in favour of the immature and less structurally sound α1α1α2 network in Alport syndrome may also lead to an extracellular matrix which is more susceptible to biomechanical strain in the renal basement membrane. 15 Correspondingly, this could lead to schitic changes through vitreomacular traction or degeneration of the retinal architecture.
It is worthwhile noting the recurrent bilateral corneal epithelial erosions in this patient, an example of Alport syndrome as a disorder of the selected basement membranes in another structure in the eye, which progressed at the same time as the schitic changes in the retina.
Summary
Alport syndrome can present with an array of retinal findings, which are believed to represent a dysfunctional Type IV collagen in the ILM and Bruch’s membrane. The availability of skilled ophthalmic examination, OCT and retinal photography allows for safe, rapid, inexpensive and accepted methods of acquiring these findings and diagnosing this syndrome. This can be particularly useful when genetic testing is unavailable or if results are not conclusive.
This is the first known report of a triad of features which include the ‘stair-case’ fovea, choroidal thinning and peripheral retinoschisis. Identifying cardinal retinal features in Alport syndrome can help for diagnosis, to detect treatable complications such as macular hole, and to identify the risk of early-onset renal failure.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This supplement and the meeting on which it was based were sponsored by Novartis (tracking number OPT19-C005). Novartis did not contribute to the content and all authors retained final control of the content and editorial decisions. Novartis have checked that the content was compliant with the Association of the British Pharmaceutical Industry Code of Practice.
Informed consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.