
Abstract
Purpose:
To describe the unusual presentation, diagnosis, and clinical course of an early-onset X-linked infantile retinoschisis
Case report:
A 6-month-old infant presented with strabismus and poor fixation. After the detection of bilateral intraretinal hemorrhage and diffuse dystrophic retinal pattern at indirect ophthalmoscopy, the patient received a complete evaluation under anesthesia. Retinal wide-field imaging, spectral domain optical coherence tomography, and electroretinogram were performed and revealed a retinoschisis involving the posterior pole and the inferior periphery in the right eye. In the left eye, an inferior retinal detachment extending to the macula was detected. Blood sample and genetic counseling were required in the strong suspicion of an inherited retinal dystrophy. Genetic tests confirmed the diagnosis of X-linked retinoschisis (RS1 gene mutation). After consultation with a pediatric vitreoretinal surgeon, a wait and see strategy was chosen. The follow up visits showed a surprisingly good natural course of the disease.
Conclusion:
X-linked retinoschisis is a well-known inherited retinal disease potentially affecting young children as early as 3 months old. In this case, the stunning presentation (diffuse retinal pigment epithelium dystrophic changes resembling a macular dystrophy) and the positive course of the disease (resolution of macular retinal detachment in the left eye and stability of schisis in the right eye) arise some interesting considerations about the necessity of an early surgical treatment.
Keywords
Introduction
Infantile retinoschisis is a relatively rare retinal disease with an X-linked recessive pattern of inheritance. 1 Consequently, it affects young males and has females as carriers, and should be taken into account in the differential diagnosis of retinal thickening in young children. 2
One of the most studied and associated genetic abnormalities is mutation in the RS1 gene (located in Xp22.13) that encodes for retinoschisin, a protein integral to the retina for cellular adhesion and tissue stability. 3
The disease manifests with an early-onset retinal disorder where the retina separates into different layers and may also detach, even through progression to true rhegmatogenous retinal detachment (RRD) is unusual. 4
The inner leaf may be notably fragile because the splitting in the retina is at the level of the ganglion cells, with extended tears between the blood vessels. Vitreous hemorrhages can also be observed.5,6 The resolution of the macular schisis with a recovery of the foveal dip has been described in rare patients after pars plana vitrectomy (PPV). 7
Case description
We describe a case with an early-onset retinoschisis and a severe presentation. The patient, 6 months old, was referred to University Eye Clinic of San Giuseppe Hospital, Milan, Italy, for the evaluation of strabismus and poor fixation. A complete standard pediatric ophthalmology consultation was planned with indirect ophthalmoscopy fundus examination, posterior pole retinal imaging, and B-scan for evaluating the retinal periphery. In consequence of the detection of a bilateral intraretinal hemorrhage and diffuse dystrophic retinal appearance OU, the patient was scheduled for evaluation under anesthesia (EUA) and pediatric vitreoretinal consultation for management and differential diagnosis in such difficult case.
During EUA, dilated fundus wide-field retinal imaging was performed (Ret Cam 3; Clarity Medical Systems Inc., Pleasanton, CA, USA) in association with hand-held spectral domain optical coherence tomography (SD-OCT) (I-Vue; Optovue, Fremont, CA, USA). Due to positive familiar history of late-onset retinoschisis and the suspect of early-onset vitreoretinal dystrophy, standard electroretinogram (ERG) with hand-held Ganzfeld (Retimax; CSO, Firenze, Italy) was performed with abbreviated ISCEV (International Society for the Clinical. Electrophysiology of Vision) protocols in sedated pediatric patient.
Exhaustive workup with multimodal imaging is mandatory in evaluating early-onset retinal dystrophies with suspect of retinal detachment (RD) since the differential diagnosis could include severe clinical pictures as familial exudative vitreoretinopathy (FEVR) and NDP-related retinopathies such as classic Norrie disease. Nevertheless, peripheral avascular retina is one of the main signs in FEVR and classic Norrie disease could have a more severe presentation with total RD, shallow anterior chamber, elevated intraocular pressure, and cataract as the presenting landmarks.
Informed consent for publishing these findings and for image publication was gathered from parents and Institutional Ethic Committee approval was obtained.
The objective examination revealed an unremarkable anterior segment OU and normal intraocular pressure (IOP) value with an iCare rebound tonometer. Refraction was slightly hyperopic OU (+4.00 D), and left eye (LE) esotropia with poor fixation was remarked. Dilated fundus examination revealed the presence of a diffuse retinal pigment epithelium (RPE) dystrophic change in the posterior pole in the LE and in the middle periphery in both eyes. A peripheral, inferior schisis cavity was evident with a huge outer layer break and pigmented edges just beyond the inferior arcades in the right eye (RE). In the LE, a bullous RD along the inferior arcade (Figure 1) was noted with a suspect threat to the posterior pole. The SD-OCT revealed a clear schisis appearance of inner retinal layers in the posterior pole in the RE (Figure 1); in the LE, the inferior bullous RD was in continuity with a shallow RD involving the macula area where the schisis cavity collapsed (Figure 2). A bilateral retinal hemorrhage within the schisis cavity was present allowing, however, detailed fundus examination.
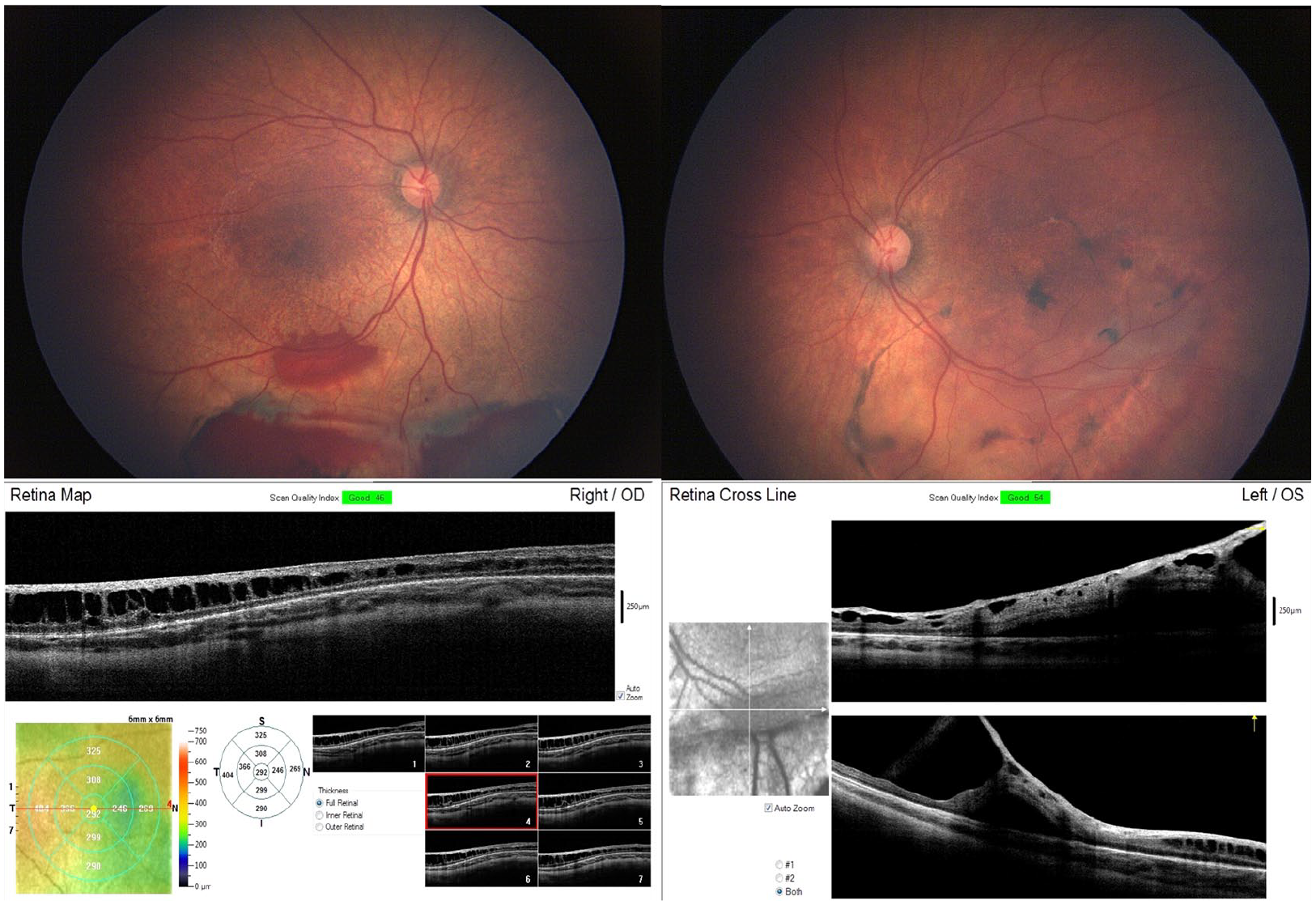
Upper right: A fundus image of the RE shows a large schisis just beyond the inferior arcades. The large hemorrhage within the schisis is clearly visible. Bottom right: The SD-OCT scan shows the schisis with the splitting at the level of the ganglion cell layer. Upper left: A fundus image of the LE shows a bullous cavity of combined schisis and retinal detachment (RD) along the inferior arcade and focal RPE hyperplasia delimiting the area of RD. Bottom left: The SD-OCT peripheral scan along the inferior arcade shows the presence of combined area of retinoschisis and localized retinal detachment.
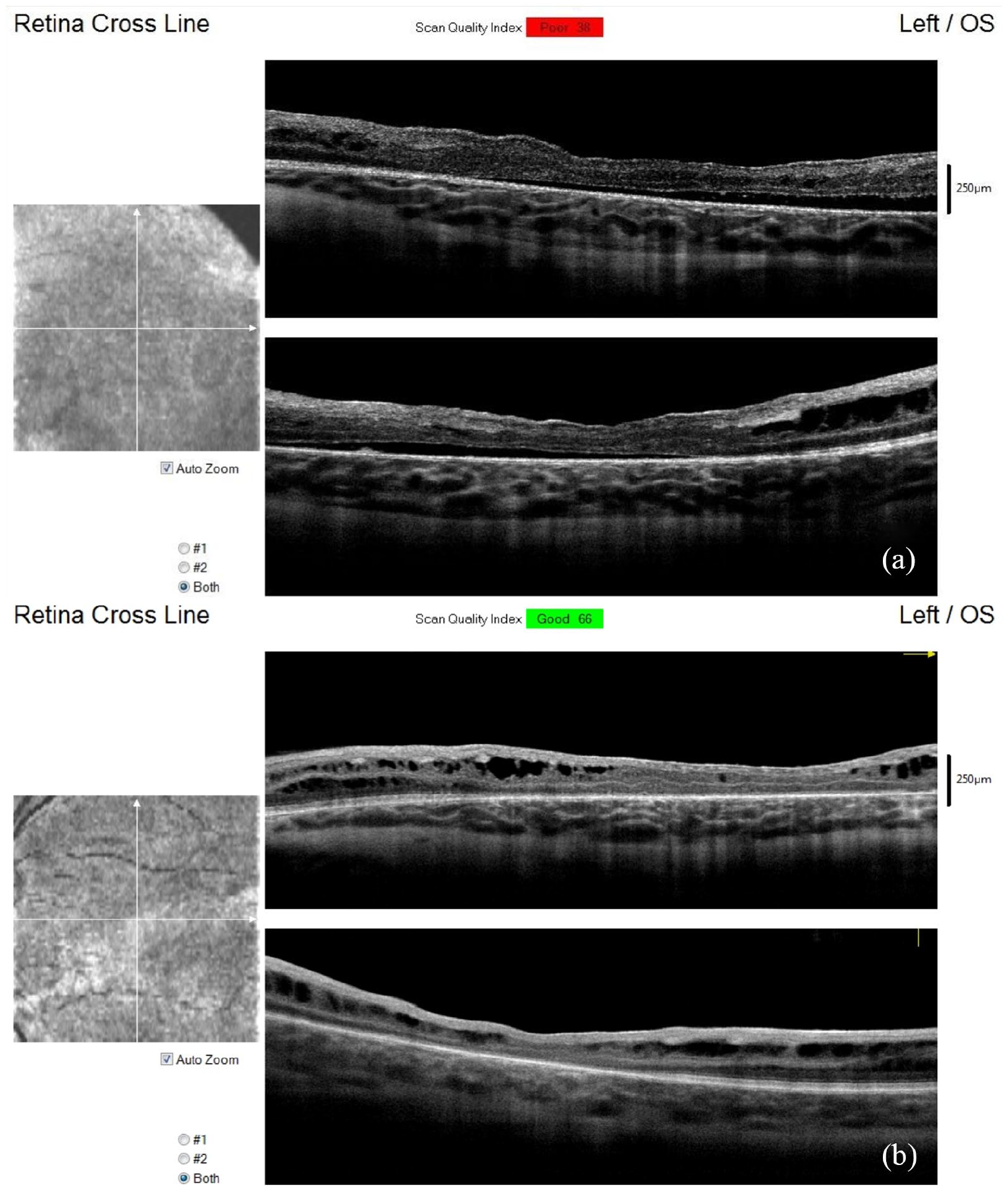
The SD-OCT of the LE shows a shallow retinal detachment involving the macula. The schisis cavity is collapsed (a). The SD-OCT 1 year later shows spontaneous resolution of retinal detachment. Some schisis cavities relapsed (b).
The standard ERG, although with reduced b-wave amplitude, possibly related to anesthetic drugs, revealed a waveform consistent with an “electronegative ERG.”
Since the strong suspect of an inherited early-onset retinal dystrophy arose from EUA, we decided not to operate the patient and proceed with a blood sample and a genetic counseling aimed to define a molecular etiology of the phenotype.
We followed the evolution of the case and scheduled a new EUA 3 months later with SD-OCT and retinal imaging to assess the natural history. EUA demonstrated a stability of the retinal presentation and a resorption of intraschisis hemorrhage, and OCT scans evidenced a resolution of LE shallow RD in the posterior pole and the presence of a stable and localized peripheral bullous RD (Figure 3).
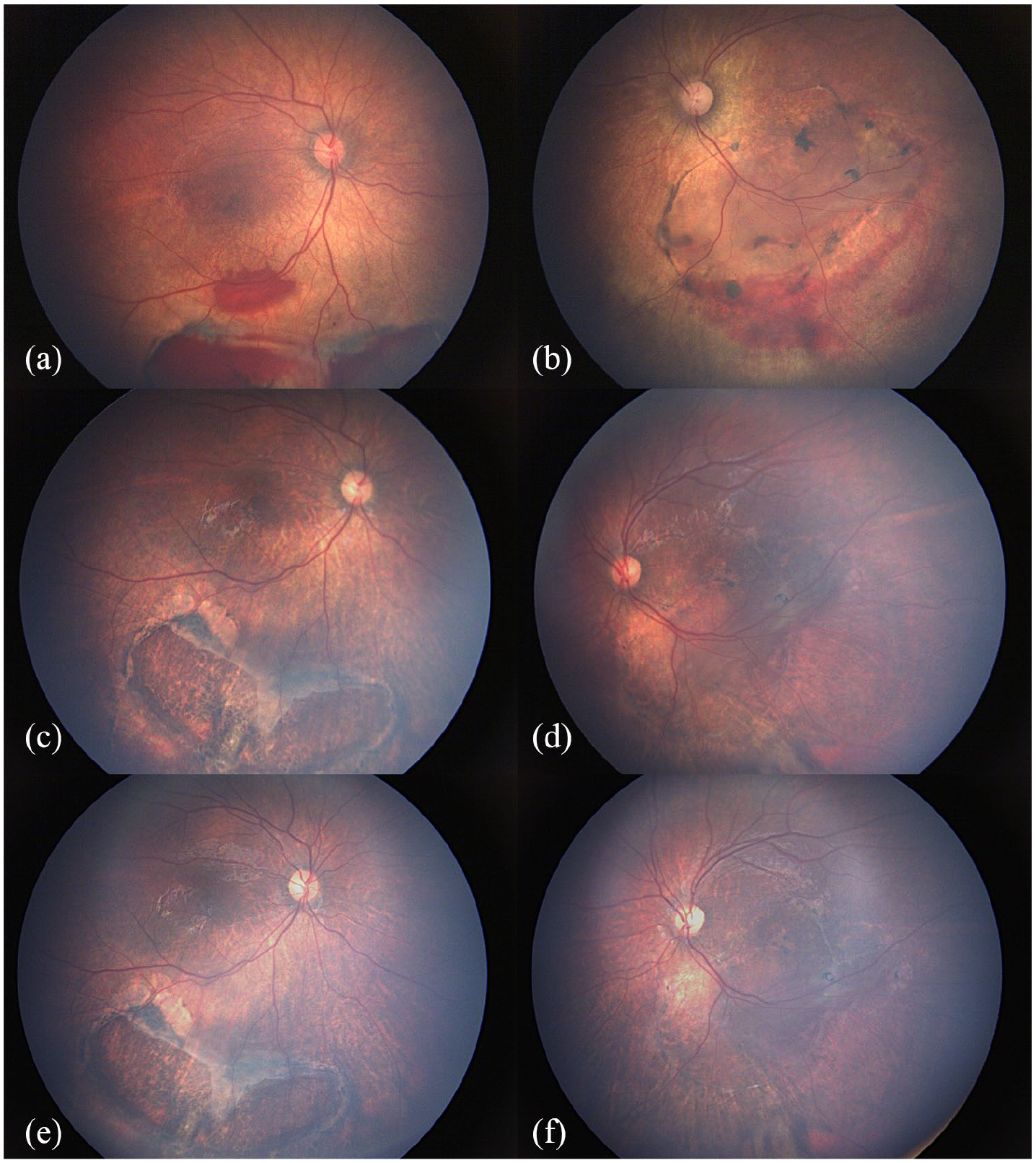
Fundus image (a, b) at baseline examination, (c, d) at 3 months, and (e, f) at 1 year of follow-up: intraschisis hemorrhage is reabsorbed. A peripheral bullous RD is reduced although visible along the infero-temporal arcades in the LE.
Fixation and following were maintained in both eyes and we followed clinically the case with an orthoptic treatment to minimize the effect of amblyopia in the LE.
After 1 year, we planned a new EUA with SD-OCT and retinal imaging: the situation was unchanged, no relapse of RD in the posterior pole, stable bullous RD, and pigmentation of outer break edges in the LE.
Genetic evaluation
Firstborn of a non-consanguineous couple, both parents were negative at ophthalmic evaluation, but the maternal grandfather recently had diagnosis of suspect retinoschisis, with reduced visual acuity.
To obtain a pregnancy, the parents previously underwent two IVF (in-vitro fertilization) cycles with no success, after which they had a spontaneous conception. Pregnancy was normal, as was noninvasive prenatal testing (NIPT) screening for most common aneuploidies. The boy was born at 41 + 3 weeks of gestation, weighed 2840 g, and had a 5I-5V Apgar because of breathing distress that was solved with bronchial aspiration. In the first days, he showed episodic bradycardia that spontaneously interrupted after few days; Holter evaluation failed to detect conduction anomalies and echocardiography showed patent ductus arteriosus (PDA), spontaneously resolved in a few months.
The boy was breastfed for 4 months and had a normal weaning. He acquired sitting position at 6 months and walking at 12 months. Language evolution was regular.
In the first clinical examination (7 months), he was 68 cm long (>25°cnt) and weighed 7.770 kg (>10°cnt), occipito-frontal circumference (OFC) was 43.4 cm (25°cnt), and he had no dysmorphic signs, except mild hypotelorism with epicanthic folds, smooth philtrum, and bowed auricles.
Molecular testing of genomic DNA from the peripheral blood evidenced a hemizygous mutation in the RS1 gene, c.544C>T (p.Arg182Cys), already reported in the literature, 8 allowing to confirm the diagnosis of early-onset X-linked retinoschisis (XLR). The mutation was detected also in the maternal grandfather.
Conclusion
We reported a case of severe, early-onset, XLR already described in the literature as possibly manifesting as early as 3 months. Our case suggests that the observation and follow-up is a valuable option in XLR despite the fact that good results could be achieved with surgical approach. 9 Retinal hemorrhage is one of the most frequent complications of the disease and usually resolves spontaneously. 2 Nevertheless, the detection of an RD in a child affected by XLR is often a major challenge with indirect ophthalmoscopy alone and differential diagnosis is very difficult without multimodal, hand-held, imaging to phenotype the cases and choose the most suitable therapeutic pathway.
Although various different treatment modalities have been previously described for managing RD in XLR, the choice of approach depends mainly on disease staging. Surgical interventions have been so far the most effective management option available for patients with advanced disease. Small-gauge vitreoretinal surgery may play a role in the management of complications (i.e. non-clearing vitreous hemorrhage) or in cases of tractional RD or macular edema sustained by epiretinal membranes (ERMs). Surgery could be the primary management when there is a risk of angle closure glaucoma in total bullous RD immediately posterior to the lens.
The stunning presentation of this case, and the natural course of the disease until today, arise some consideration in the management of early-onset XLR. First of all, these patients need a detailed clinical evaluation from trained pediatric ophthalmologists and from vitreoretinal surgeons with pediatric expertise in order to diagnose and approach correctly the case, plan the best follow-up, and optimize the rehabilitation treatment to maximize the functional outcome.
Our patient did not show typical cysts in inner retinal layers of the LE where RD was present: some authors argued that schisis cavities collapse in the presence of RD because the centripetal forces are not balanced by centrifugal forces due to correct adhesion of photoreceptors to RPE. 10 In our case, the resorption of RD did correlate with a partial restoration of the classical schisis pattern in the posterior pole: this observation could confirm the previous hypothesis, even if more factors could contribute to it.
The last consideration is about the management of retinal findings: even in the presence of a big break in the outer wall, a close follow-up could be the right choice if the inner wall is intact and no progression to RD is noticed.
Footnotes
Authors’ note
Prof. P.N. confirms to be an associate editor of this journal and was not involved in the peer review process for this paper.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.