
Abstract
Introduction:
The ocular presentation of paraneoplastic pemphigus (PNP) has rarely been reported in the literature. In this report, we describe a 61-year-old male presenting with eruptive skin lesions associated with underlying non-Hodgkin’s lymphoma who had rapid progressive corneal perforation with secondary endophthalmitis in the setting of PNP.
Case description:
A 61-year-old male presented to the emergency department complaining of skin eruption mimicking Stevens–Johnson syndrome, which was later found to be related to PNP. Initially, the patient complained of progressive ocular surface dryness in both eyes. Meanwhile, he developed mild pain in the right eye associated with blurry vision in both eyes and was managed with lubricants and topical antibiotics. A few days later, he was found to have corneal perforation with features suggestive of left endophthalmitis with possible early panophthalmitis. Intravenous antibiotic was administered, and primary evisceration of the left globe was performed. Histopathology revealed acute necrotizing keratitis and endophthalmitis. Vitreous analysis showed numerous gram-negative bacilli and a positive culture of Morganella morganii. The patient continued to be managed with frequent lubrications and punctual plugs in the fellow eye during the follow-up period.
Conclusion:
We describe the first case of endophthalmitis developing secondary to PNP-induced corneal melting and perforation. Anticipating unusual infectious sequelae in the setting of PNP might be warranted to actively detect and successfully manage dry eye disease before devastating complications develop.
Keywords
Introduction
Paraneoplastic pemphigus (PNP) is a rare autoimmune multiorganic heterogeneous mucocutaneous syndrome that is, fundamentally associated with an underlying neoplasia. It characteristically presents initially with painful oral and skin erosions with or without eruptions. 1 Other sites targeted by the disease include the esophagus, nasopharynx, lung, stomach, colon and ocular tissue. Ocular manifestations in PNP include forniceal shortening, symblepharon formation, bilateral conjunctivitis with hyperemia and corneal erosions.1,2 Corneal ulceration and subsequent perforation rarely occur but are possibly explained by the nature of PNP pathogenesis. We describe the clinical and microbial characteristics of acute necrotizing keratitis with subsequent endophthalmitis in a patient presenting with PNP.
Case description
A 61-year-old male presented to the emergency department complaining of skin eruption that was thought to be secondary to Stevens–Johnson syndrome with mucosal involvement. He was screened for malignancy and was found to have incidental retro-peritoneal lesions. Computed tomography (CT)-guided biopsy was suggestive of low-grade B-cell non-Hodgkin’s lymphoma (NHL). The diagnosis of PNP was established, and the patient was started on oral steroids (1 mg/kg) and then intravenous immunoglobulin (IVIG), which showed an improvement in terms of skin eruptive lesions but not mucosal lesions. In addition, the patient was referred to the oncology centre and was started on the standard regimen of rituximab plus cyclophosphamide, vincristine and prednisone (R-CVP). Before being discharged, he was seen by the ophthalmology team and was found to have a dry ocular surface with a visual acuity of 20/25 in both eyes.
The patient started to complain of intermittent dry eye symptoms, which progressed severely within a period of 3 months. The patient presented to the emergency room (ER) with severe pain in his left eye; however, the vision was preserved at that time, so he was managed with heavy topical lubricants. Two days later, the patient presented again to the ER with high-grade fever associated with severe pain, lid swelling and loss of vision in the left eye. Visual acuity measurements were 20/50 and no light perception for the right and left eyes, respectively. The extraocular muscle motility was full in the right eye, whereas restricted motility was observed in the left eye. Slit lamp examination of the right eye showed severe confluent superficial punctate keratopathy with filamentary keratitis (Figure 1(a) and (b)). Ophthalmic examination of the left eye revealed a severely tender, erythematous and edematous upper eyelid with severe conjunctival injection and chemosis. The cornea showed central thinning with apparent perforation and iris prolapse. The anterior chamber collapsed with an underlying hypopyon. A B-scan of the left eye revealed significant retino-choroidal layer thickening with dense vitreous opacities and membrane formation, with periscleral fluid collection suggestive of early panophthalmitis.
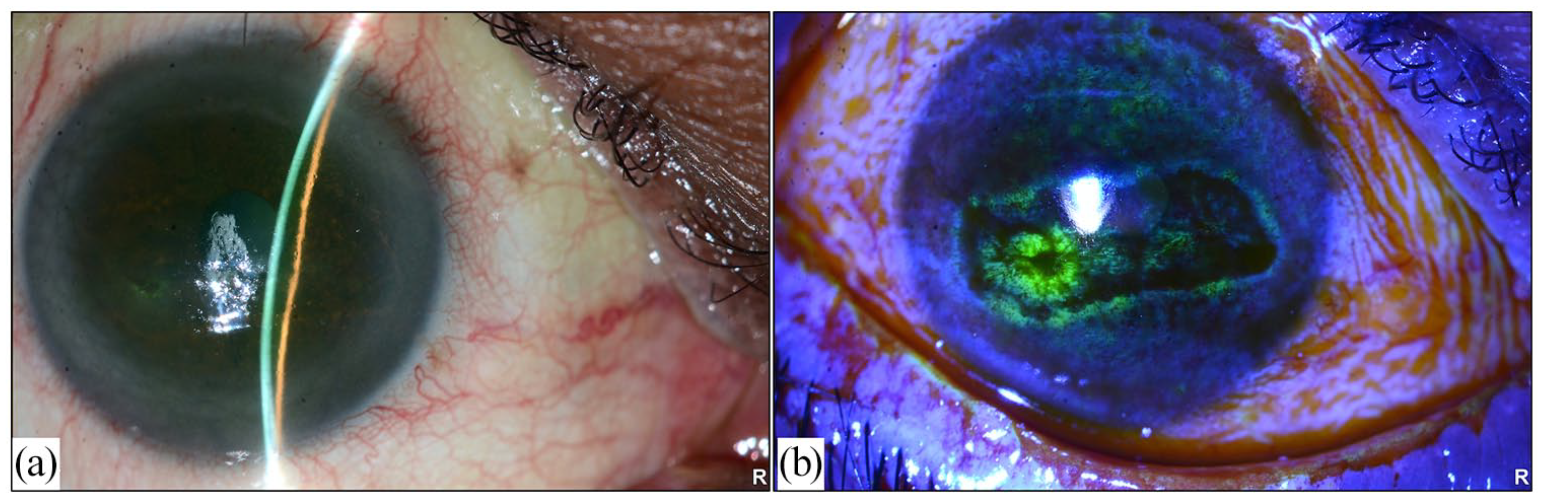
(a) Slit lamp photo of the right eye showing loss of corneal luster and significant dry ocular surface and (b) right eye photo with fluoresceine stain showing conjunctival staining and confluent superficial punctate keratopathy and filamentary keratitis.
The clinical diagnosis of rapidly progressing severe endophthalmitis secondary to corneal perforation was established upon admission. Blood workup was ordered, and intravenous 600 mg clindamycin and 2 g ceftriaxone were administered. In an effort to avoid progression to panophthalmitis, the patient was scheduled for urgent primary evisceration versus enucleation of the left globe depending on the intraoperative findings to avoid intracranial or systemic spread of the infection. Intraoperatively, the decision for evisceration was made based on the relative sparing of the sclera. Histopathological evaluation of the eviscerated eye contents revealed acute necrotizing keratitis with corneal ulceration and perforation. In addition, it showed severe uveal tissue necrosis and acute inflammation with intraocular collection of acute inflammatory exudates consistent with endophthalmitis of the left globe (Figure 2(a)). Gram staining indicated a collection of gram-negative bacilli within the vitreous (Figure 2(b)). The culture showed bacterial growth, and Morganella morganii species was identified. The postoperative period was unremarkable for the operated eye. However, the right eye developed a spontaneous large corneal epithelial defect that was managed with frequent lubrication, punctal plug placement and daily follow-up until complete healing of the epithelial defect. Three consecutive blood and urine cultures were taken and showed negative cultures, ruling out the possibility of endogenous spread of the organism. The patient was then shifted to intravenous cefepime 2 g every 8 h for 3 days. Afterward, the patient was discharged on oral ciprofloxacin 750 mg twice a day for 14 days. One week postoperatively, ophthalmic examination of the right eye showed severe superficial punctate keratopathy with no corneal epithelial defect. Currently, the patient is being followed up by a hemato-oncologist and is continuing the sixth cycle of the R-CVP regimen.
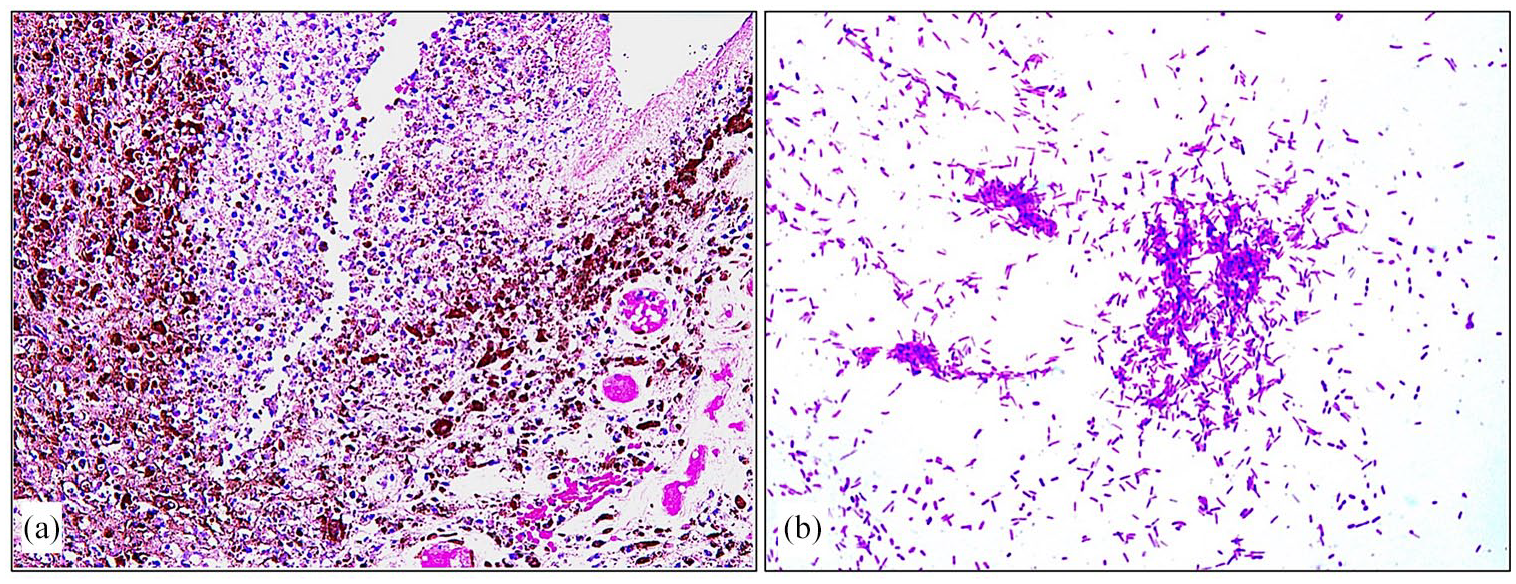
(a) Uveal tissue necrosis and acute inflammation (original magnification ×200 Hematoxylin & Eosin) and (b) collection of gram-negative bacilli within the vitreous consistent with bacterial endophthalmitis (original magnification ×1000 oil Gram stain).
Discussion
The term ‘paraneoplastic pemphigus’ (PNP) falsely implies only one aspect, that is, skin involvement, whereas the more accurate term ‘paraneoplastic autoimmune multiorgan syndrome (PAMS)’ has been increasingly used recently, as it encompasses a more comprehensive description of this entity. Although there are no current reliable epidemiological estimates of PNP, approximately 500 cases have been reported worldwide during the past three decades. 1 The majority of PNP is caused by hematologic neoplasms or lymphoproliferative disorders, with non-Hodgkin’s lymphoma being the most common neoplasm reported (38.6%), followed by chronic lymphocytic leukemia (18.4%) and Castleman’s disease (18.4%).1,2
Screening for occult malignancy in PNP is not yet standardized but might be essential, especially in cases not responding to immunosuppressive therapy. 2 In 30% of cases, PNP precedes the diagnosis of malignancy, which is similar to our report.2,3 Therefore, we screened our patient with pan-CT, detected the source of the tumour and finally established a diagnosis of non-Hodgkin’s lymphoma.2,3 PNP typically affects adults aged between 45 and 70 years and accounts for <5% of all pemphigus cases. 2 Our patient initially presented with an eruptive skin lesion mimicking pemphigoid vulgaris associated with severe eye dryness. Therefore, identifying skin lesions with high-risk features would be helpful to guide the diagnostic and management plan.
Several differentiating diagnostic aspects exist between paraneoplastic pemphigoid and pemphigus vulgaris.1,4 First, PNP is a multiorgan syndrome affecting ocular, pulmonary and gastrointestinal mucosa with or without neuromuscular involvement, unlike pemphigus vulgaris, which is only limited to mucocutaneous epithelium. 4 Additionally, PNP develops autoantibodies against plakophilin 3 and desmoglein 1–3 with characteristic findings in immunochemical and histopathological testing, which are not apparent in pemphigus vulgaris. In contrast to pemphigus vulgaris, which is benign with a favourable prognosis, PNP is basically associated with underlying malignancy with a poor response to immunosuppressive agents and a mortality rate of 90% within the first year following diagnosis.1,2,4
The most common ocular presentation of PNP is bilateral chronic cicatrizing conjunctivitis. 5
Other ocular manifestations in PNP include forniceal shortening, symblepharon formation and corneal erosions. 4 The disease in our case was asymmetrically bilateral, where the left eye was significantly more affected, with corneal erosion that progressed rapidly to corneal thinning and perforation. The rapid progression led to the introduction of opportunistic microorganisms to the intraocular structures, resulting in necrotizing keratitis and endophthalmitis, which was the most devastating ocular complication in our patient. We believe that our early intervention played a role in hindering the progression to full-blown panophthalmitis. Limbal stem cell deficiency (LSCD), as a sequela from long-term ocular pemphigoid, could explain the progression of persistent epithelial defects into ulceration and corneal perforation. For this reason, Tam et al. 5 recommended providing adequate lubrication and corneal protection to prevent any irreversible blinding complications. Another potential cause for LSCD in our case was the use of systemic chemotherapy, as observed previously with some chemotherapies. 6 However, the severity, chronicity and overall clinical scenario of this bilateral cicatrizing keratoconjunctivitis despite the use of heavy lubricants would support PNP as an etiology.
Morganella morganii is an opportunistic gram-negative bacillus classified under the family of Enterobacteriaceae pathogens and usually resides in the human intestinal tract as a normal flora. Morganella morganii causes biliary and urinary tract infections and rarely contributes to ocular infection. 7 Few reports have addressed Morganella morganii as a cause of panophthalmitis, with the first reported case of acute postoperative Morganella morganii panophthalmitis occurring after vitrectomy. 8 Based on histopathological examination and vitreous culture results, our patient developed secondary endophthalmitis following corneal perforation in the absence of an endogenous source, suggesting direct spread of the organism intraocularly. The patient’s age, medical history of hematologic neoplasm and biliary disease and frequent use of antibiotics were all additional predisposing factors for the acquisition of Morganella morganii nosocomial infection in our case. 7
As the patient was immunocompromised with a clear focus of infection, we initiated empiric antibiotic treatment postoperatively despite apparent clinical stability and negative blood and urine cultures. Similar to other Enterobacteriaceae species, Morganella morganii tends to develop resistance against β-lactam antibiotics. 8 Therefore, a trial of intravenous cefepime 2 g, a fourth-generation cephalosporin, was given with oral ciprofloxacin to avoid the risk of β-lactamase cross-resistance. Fortunately, our patient did not develop bacteremia, where three consecutive blood cultures were negative, and responded well to antimicrobial coverage. Although the patient had already lost one eye and the prognosis was unfavourable in terms of associated systemic comorbidities, managing dry eye disease remained a major determinant of the patient’s quality of life. Applying a stepladder approach, we administered IVIG to control the activity of pemphigoid lesions, as the case was nonresponsive to the initial conventional steroid therapy.
Conclusions
This is the first report of the development of bacterial endophthalmitis with impending panophthalmitis secondary to corneal perforation in the setting of PNP. A lower threshold to detect corneal involvement, anticipate infectious complications and treat them aggressively is warranted when approaching a patient with PNP.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Informed consent was obtained from the patient for the anonymous use of data.