
Abstract
Background:
Von Hippel-Lindau syndrome is a rare autosomal dominantly inherited multisystemic oncologic syndrome, presenting predominantly with angiomatosis in embryologically similar neurologic tissue such as retina, cerebellum and adrenals. Retinal hemangioblastomas are the hallmark ophthalmic finding. In this case report, we describe the importance of timely diagnosis, thorough systemic examination and treatment of bilaterally asymmetrical retinal hemangioblastomas in a young adult male.
Case presentation:
A 31-year-old male presented with painless diminution of vision in both eyes, associated with eyestrain and headache. Multiple asymmetric retinal lesions and dilated feeder vessels were noted on ophthalmoscopic examination and confirmed by fluorescein angiography to be retinal hemangioblastomas. Comprehensive systemic examination revealed cerebellar hemangioblastomas and multiple pancreatic and renal cysts. Treatment of retinal lesions was done by combination therapy of argon laser photocoagulation and cryopexy, which lead to a good visual outcome. Subsequently, neurosurgical resection of cerebellar hemangioblastoma proved to be lifesaving for the patient.
Conclusion:
RHBs are the earliest, easiest and the most frequently detected manifestation of VHL. Identification of ocular manifestations play a pivotal role in early diagnosis and timely intervention in VHL syndrome, thereby significantly reducing associated morbidity and mortality. Therefore, an ophthalmologist’s role is crucial in the management of these potentially deadly tumours.
Introduction
Von Hippel-Lindau syndrome (VHL) (OMIM number 193300) is a rare autosomal dominantly inherited multisystemic oncologic syndrome associated with germline mutation of the VHL gene. The most common manifestations of VHL include hemangioblastomas of the retina and central nervous system (CNS), kidney cysts, renal cell carcinoma (RCC), pheochromocytoma, pancreatic cysts and primitive neuroendocrine tumours (PNETs). The hallmark ophthalmic finding in VHL syndrome is retinal hemangioblastoma (RHB) with a prevalence between 45% and 65%. 1 While the median age of onset is around 21 years, the probability of developing retinal hemangioblastoma increases as age progresses. Although, retinal hemangioblastomas develop as small solitary tumours that may remain stable, in up to 50% of patients there can be bilateral multiple lesions, which tend to increase in size over time. 2 Secondary tumour effects like intraretinal and subretinal exudation may occur, leading to significant visual loss. We present this case report to highlight the importance of identifying the ocular manifestations of VHL as well as to emphasize that timely intervention can significantly reduce morbidity and mortality associated with the disease.
Case presentation
A 31-year-old man presented with a painless, decrease of vision in both eyes, associated with eye strain and headache for the past 3 weeks. His best-corrected visual acuity was 0.30 logMAR (6/12 by Snellen) in the right eye and 0.20 logMAR (6/9.5 by Snellen) in the left. Right eye fundus examination with 90-dioptre noncontact lens revealed significant macular oedema. In addition, on indirect ophthalmoscopy, two small, well-circumscribed sessile orange-red appearing lesions were noticed in the superior retina. (Figure 1(a)) Left eye posterior pole examination was unremarkable. However, on indirect ophthalmoscopy, one large (6–7 mm) globular reddish lesion bulging into the vitreous was noticed in the superior retina along with a dilated feeding vessel and tortuous draining vein, characteristic for RHB. Five small (<1.5 mm) similar lesions were also noticed in the periphery. (Figure 1(b)) Fluorescein angiography showed early yet progressive hyperfluorescence, with late leakage of fluorescein into surrounding retina in both eyes. (Figure 1(c) and (d)) Comprehensive systemic evaluation, including magnetic resonance imaging (MRI) of brain, renal ultrasonography and computed tomography of abdomen was undertaken. It revealed cerebellar hemangioblastoma (Figure 2) and multiple pancreatic and kidney cysts. (Figure 3) Detailed family history revealed that maternal grandfather and uncle, both had suffered from advanced unilateral vision loss and died from brain and kidney tumours; no significant family history was elicited from the mother. Based on the clinical and investigational findings, the diagnosis of VHL syndrome was made in this patient.
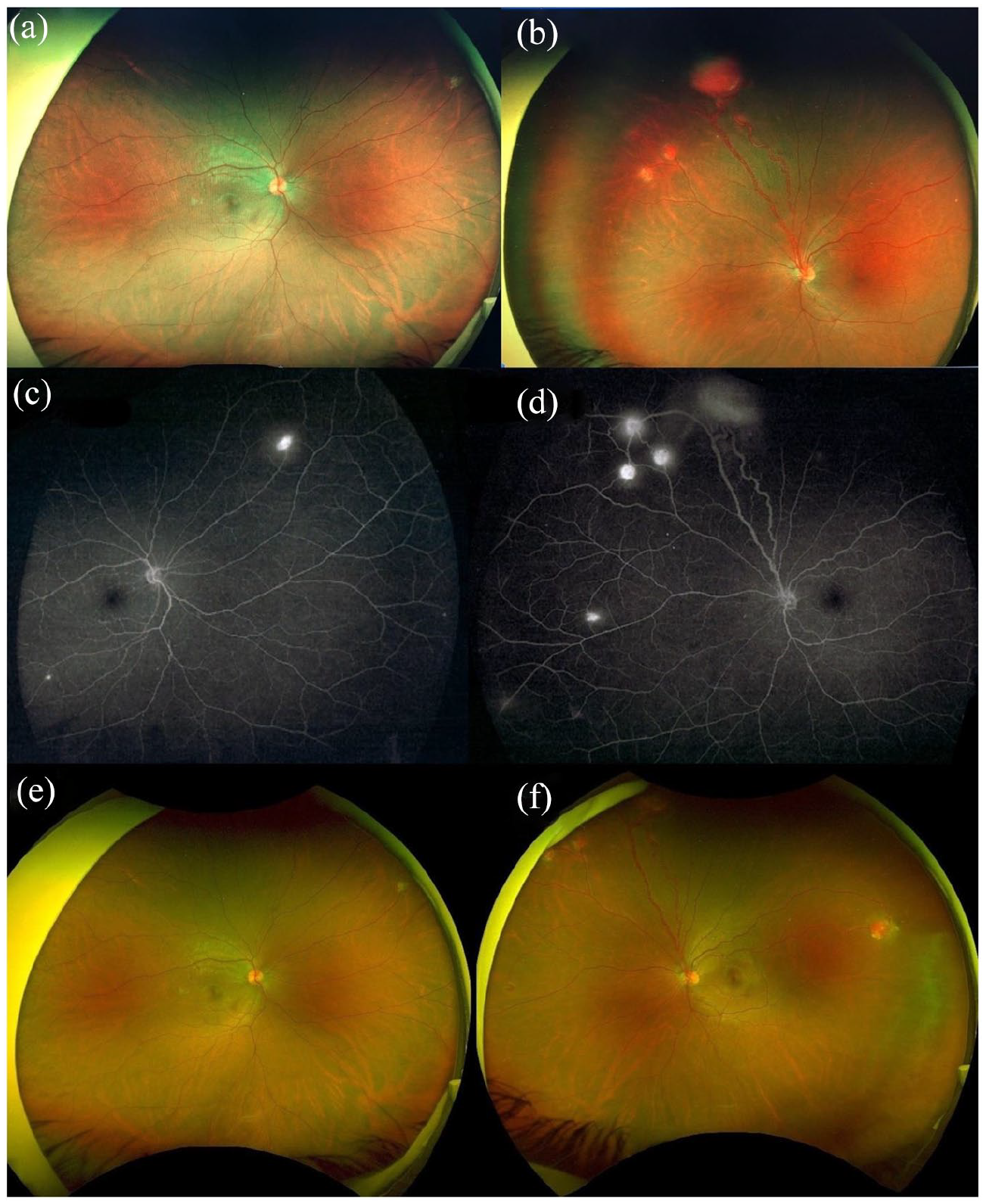
Fundus photography by ultra-wide field (UWF) Optos™ imaging shows multiple retinal hemangioblastomas in right (a) and left (b) eye. Fluorescein angiography (c, d) depicts homogeneous capillary filling of tumour and fluorescence of dilated feeder arteriole and venule. Post-treatment, clinical photograph by UWF-Optos shows bilateral RBH regression (e, f).
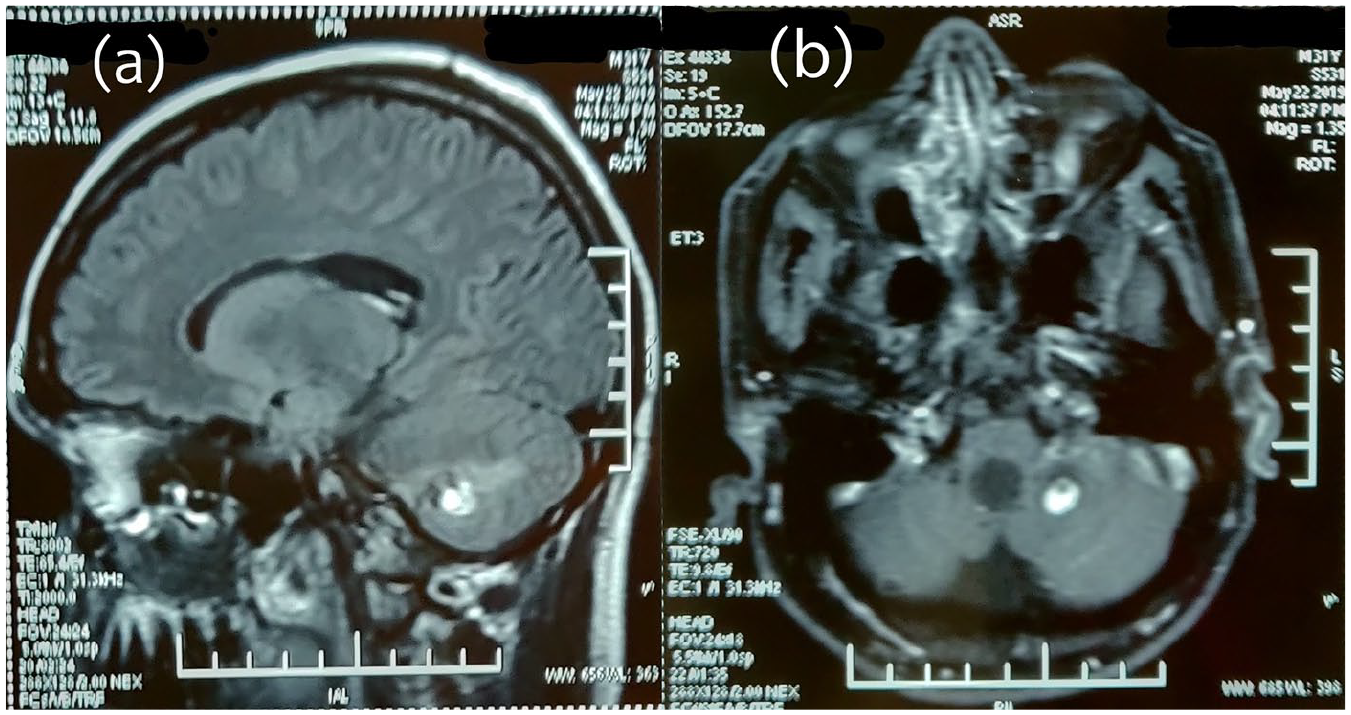
MRI brain, sagittal (a) and axial (b) sections reveal a sharply demarcated homogenous mass depicting both solid and cystic components, suggestive of cerebellar hemangioblastoma.
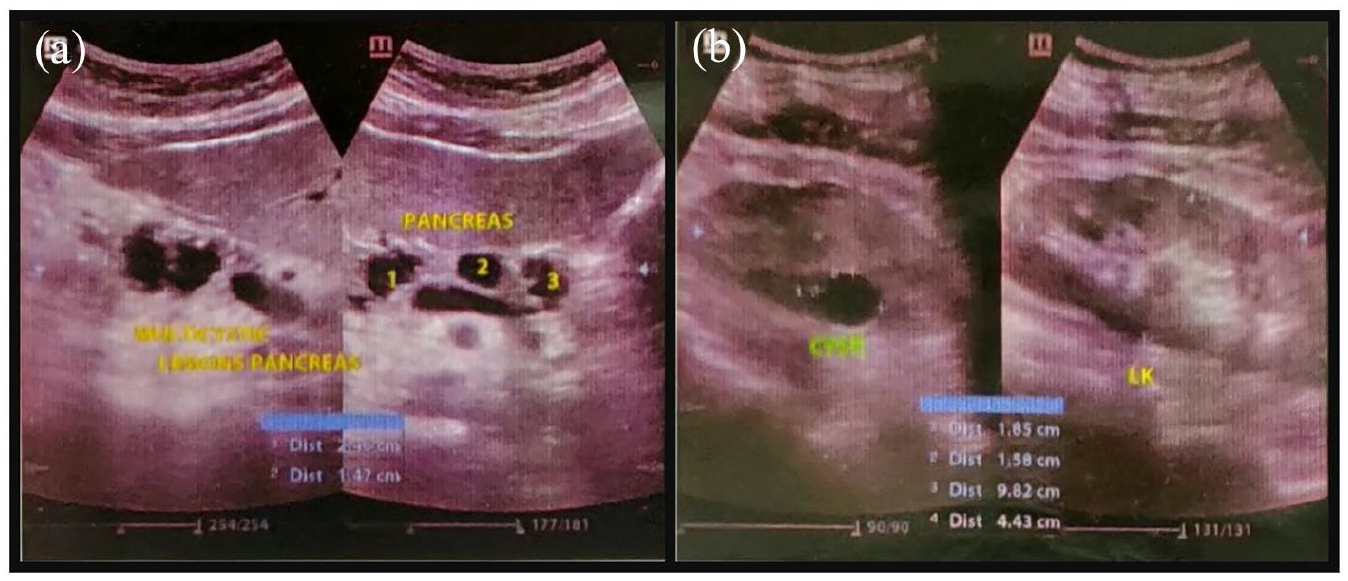
Abdominal ultrasonography scan shows multiple cysts in the head of pancreas (a) and left kidney (b).
Transpupillary Argon laser photocoagulation was used to treat the small (⩽1.5 mm) hemangioblastomas in both eyes and as for the large RHB in the left eye, combination therapy of laser photocoagulation and cryopexy was used, over three sittings. Large size (500 microns), low intensity (250 mW) and long duration (0.5 s) laser burns were applied directly on the RHB surface in both eyes, after photocoagulating and occluding the feeding artery. Cryopexy was done using a double freeze-thaw technique under indirect ophthalmoscopy. Complete regression of RHBs resulted in formation of chorioretinal scars. After 3 months of follow-up, macular oedema in the right eye regressed and no new RHB was observed in either eye (Figure 1e,f). His visual acuity improved to 0.1 logMAR (6/7.5 by Snellen) and 0.2 logMAR (6/9.5 by Snellen) in the right and left eye, respectively. In the first year of observation, patient also underwent neurosurgical resection of cerebellar hemangioblastoma and is doing well on his third annual follow-up examination with no signs of recurrence.
Discussion
VHL syndrome is a rare, autosomal dominant, monogenic disease arising from mutations of the VHL gene, which predisposes to the development of specific benign and malignant tumours. The prevalence of VHL syndrome is estimated to be between 1 in 31,000 and 1 in 53,000.3,4 The VHL gene is a tumour suppressor gene, located on the short arm of chromosome 3 (3p25–26) and is expressed in all organ systems, not exclusively those affected by VHLsyndrome. 5 This gene functioning involves diverse cellular level processes like angiogenesis, proliferation, metabolism and apoptosis. Therefore, its mutation disrupts these processes and results in angiomatosis in embryologically similar neurologic tissues such as retina, cerebellum, adrenals, etc. According to the phenotypic characteristics, VHL syndrome can be divided into four phenotypic groups based on the presence of pheochromocytoma or renal cell carcinoma (type 1, type 2A, type 2B and type 2C). Pheochromocytoma is not present in type 1, but occurs with all other types. Considering his clinical findings and family history, our patient was consistent with type 1. 6
The characteristic vascular tumours of retina were first described as ‘angiomatosis retinae’ by a German ophthalmologist Eugene von Hippel in 1911. 7 Lesions are often bilateral and multiple. These are commonly located in the peripheral temporal retina, but may occur in the posterior pole and on the optic disc. 8 Coats disease, retinal cavernous haemangioma, retinal macroaneurysms and vasoproliferative tumours form the differential diagnosis. 9 Despite being slow-growing and benign tumours, RHBs can cause complications such as macular oedema, exudative and tractional retinal detachment, intravitreal haemorrhage and neovascular glaucoma, if appropriate treatment is not provided timely. 10 Fundus fluorescein angiography using ultra-widefield imaging (FFA-UWF) is a highly useful diagnostic modality as it helps in delineation, quantification and localization of RHB, characteristic of VHL syndrome. 11
Traditionally, treatment options include laser photocoagulation, cryotherapy, photodynamic therapy, transpupillary thermotherapy, plaque radiotherapy, external beam radiotherapy and vitreoretinal surgical ablation. The treatment modality is chosen based upon the location, size of lesion and related complications. Laser photocoagulation is more commonly used to treat smaller tumours, while cryotherapy is more often preferred for very peripheral tumours larger than 3 mm. 12 Hence, in this case, we chose a combination of laser photocoagulation and cryotherapy for the left eye, and started the treatment as soon as VHL syndrome was diagnosed. This approach proved effective in destroying the tumour and consequent improvement in visual acuity. The likelihood of a favourable outcome is lower with tumours located on or around the optic disc. 7 Recently, intravitreal anti-vascular endothelial growth factor inhibitors have proven useful in reducing exudation resulting from RHBs. 13
In this case, VHL syndrome was suspected based on findings of multiple RHBs. The presence of renal, pancreatic cysts, cerebellar hemangioblastoma and a positive family history strengthened this diagnosis. However, due to lack of resources, genetic testing could not be performed, which would have confirmed the same. Patients’ relatives were informed about this genetic condition and they underwent necessary screening examination and investigations. The patient was advised against the use of tobacco products and to avoid contact sports in view of the renal and pancreatic cysts.
Conclusion
VHL is a potentially life-threatening syndrome that is seldom encountered in routine clinical practice. Involvement of various organ systems warrants a multidisciplinary treatment approach, making its overall management challenging. RHBs are the earliest, easiest and the most frequently detected manifestation of VHL. Consequently, these characteristic eye tumours constitute diagnostic criteria of most clinically defined VHL cases.
An ophthalmologist’s role, in identifying RHBs and establishing their association with VHL, is extremely significant for the early diagnosis and treatment of these potentially deadly tumours. Ocular morbidity and mortality can thus be prevented in these patients and their relatives. The benefits of thorough systemic evaluation, history taking and screening of family members, timely intervention and an earnest effort to follow up the patient are evident in this case report.
Footnotes
Author contributions
ST played pivotal role in diagnosis and management of the patient and drafting the initial manuscript. KCK contributed by critically evaluating and refining the manuscript in its present form. KS substantially contributed to acquisition of clinical and investigative data and revising the report herein. KCK and KS have approved the submitted version and have agreed to be both personally accountable for their individual, as well as group contribution. All of us ensure that questions related to the accuracy or integrity of any part of the work has been appropriately investigated and resolved.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Requisite approval from the institutional ethics review committee was taken.
Informed consent
Informed consent for clinical and radiological data was obtained prior to drafting the manuscript.