
Abstract
Lipoid proteinosis (LP), or Urbach-Wiethe disease, is an ultra-rare autosomal-recessive disorder caused by loss-of-function variants in the ECM1 gene. It is characterized by the progressive deposition of hyaline-like material in the skin, mucosae, and central nervous system. This series describes three siblings (aged 16, 24, and 25) from Gujranwala, Pakistan, born to non-consanguineous parents. All patients presented with infant-onset hoarseness of voice and recurrent vesiculobullous eruptions that evolved into waxy papules and hyperkeratotic plaques. Physical examination revealed the pathognomonic “beaded” eyelid papules (moniliform blepharosis), pock-like facial scarring, and a thickened lingual frenulum restricting tongue protrusion. Despite shared genetics, intrafamilial variability was observed, with the male sibling exhibiting a more severe “leonine-like” facies. Neuropsychiatric symptoms, including aggression and insomnia, were noted across the series. Due to resource constraints, the diagnosis was established clinically. Management focused on symptomatic control using oral acitretin (0.5 mg/kg/day), antihistamines, and intensive topical emollients. At 12-week follow-up, patients showed significant reduction in pruritus and stabilization of skin lesions, though hoarseness and established scarring remained unchanged. This series underscores the importance of recognizing cardinal clinical triads—moniliform blepharosis, mucosal tethering, and early hoarseness—to diagnose LP in resource-limited settings where genetic or histological confirmation is unavailable.
1. Introduction
Lipoid proteinosis also called the Urbach-Wiethe disease, lipoglycoproteinosis as well as hyalinosis cutis et mucosae (due to glass like appearance of the affected skin, mucous membrane and internal organs) is a rare autosomal-recessive disease originally described as “lipoidosis cutis et mucosae” by Urbach and Wiethe. 1 This ultra rare disease was called as lipoid proteinosis because histopathologic features on skin biopsy that showed abnormal protein and lipid inclusions. LP is inherited in an autosomal-recessive manner and is caused by biallelic loss-of-function variants in the extracellular matrix protein 1 (ECM1) gene on chromosome 1q21, which encodes a glycoprotein essential for the structural integrity and homeostasis of the skin and mucosae. 2 The clinical picture may vary in patients however mouth, skin, pharynx and larynx are most affected organs. It mostly manifests as hoarseness of the voice due to laryngeal thickening from hyaline deposition. 3 Neurological manifestations may also occur due to calcifications in amygdala and temporal lobes. Yellow-waxy papules, plaques, moniliform blepharosis, inflammation of salivary glands, inability to protrude the tongue due to a thickened frenulum and verrucous lesions on extensor surfaces subject to friction for example elbows, knees, buttocks, and knuckles are commonly observed clinical manifestations. 4 There are around 400 documented cases of this disorder worldwide, with around 50 published cases demonstrating neurological and neuropsychiatric sequelae. 5 Although LP occurs worldwide, fewer than 500 cases have been reported to date; a well-recognized founder effect accounts for an unusually high prevalence in the Namaqualand region of the Northern Cape, South Africa, while elsewhere the disorder is reported most frequently from communities with high rates of consanguinity. 6 This rarity, together with its autosomal-recessive basis and wide phenotypic expressivity, makes well-characterized intra-familial series particularly valuable. Here we present a clinically diagnosed series of three affected siblings from a non-consanguineous Pakistani family. Rather than an isolated case report, this series is intended primarily as a comparative description of intra-familial phenotypic variability in LP, emphasizing the cutaneous, mucosal, and otolaryngological features that enable clinical recognition and diagnosis in resource-limited settings where histological or genetic confirmation is unavailable. 7
2. Methods
This case series was conducted at the dermatology outpatient department of a tertiary-care hospital in Punjab, Pakistan. Three siblings from a single family who presented contemporaneously with a clinically suggestive mucocutaneous and otolaryngological phenotype were identified and consecutively enrolled. A detailed history was obtained from the patients and their mother, with particular attention to the age and sequence of symptom onset, the family pedigree, and parental consanguinity. Each sibling underwent a structured clinical assessment comprising full cutaneous examination, oral and oropharyngeal inspection, examination of the eyelid margins, and a brief neuropsychiatric evaluation; findings were graded semi-quantitatively to permit direct comparison of phenotype across the three siblings.
Because dermatopathology, ECM1 sequencing, and neuroimaging were not available in this resource-limited setting, the diagnosis was established on clinical grounds and recognized phenotypic mimics were systematically excluded. All three patients received an identical management protocol based on oral acitretin, antihistamines, and topical emollient and barrier therapy, with baseline laboratory testing performed before initiation of systemic retinoid therapy. Patients were reviewed at 4-week intervals over a 12-week period, and cutaneous response, mucosal and laryngeal symptoms, pruritus, treatment adherence, and adverse effects were recorded at each visit. Written informed consent for participation and for the publication of de-identified clinical images was obtained from the adult patients and from the legal guardian of the minor.
3. Case Presentation
3.1. Patient Information and Clinical Context
Three siblings (two females aged 24 and 25 years, and one male aged 16 years) from Gujranwala, Punjab, Pakistan, born to a non-consanguineous marriage, presented to the outpatient department on 14 October 2025 with recurrent mucocutaneous lesions and hoarseness of voice since early infancy. According to the mother, all three patients developed symptoms at approximately 2–3 months of age, initially as vesiculobullous eruptions that later evolved into papules and plaques. There was no known history of similar illness in previous generations. The clustering of cases within the same family strongly suggested a genetic etiology.
3.1.1. Case 1
A. Patient Information
A 24-year-old female presented with a lifelong history of recurrent waxy papules and plaques beginning in early infancy. B. Clinical Findings
The patient developed vesicles at approximately 2 months of age that eroded and crusted following minor trauma. Over time, these lesions evolved into hyperkeratotic plaques distributed over the back, abdomen, axilla, inguinal region, elbows, knees, and buttocks. The lesions were associated with pruritus and photosensitivity.
Cutaneous examination revealed diffuse waxy papules coalescing into plaques with areas of crusting and erosion. Prominent pock-like facial scarring and exaggerated perioral and forehead lines gave a prematurely aged appearance. Moniliform blepharosis was present along the eyelid margins.
Oral examination demonstrated a thickened, short lingual frenulum restricting tongue protrusion, along with nodular infiltration of the tongue, soft palate, and posterior pharynx. Persistent hoarseness of voice was noted since infancy.
Neurological features included aggression, migraine, and insomnia. Nail findings included shiny nails and nail fold atrophy. Representative clinical findings are shown in Figure 1. Clinical findings in Case 1 (Sibling 1); representative clinical images of the 24-year-old female demonstrating diffuse waxy papules and hyperkeratotic plaques involving the trunk and extremities, along with characteristic pock-like facial scarring and exaggerated perioral lines. Moniliform blepharosis is evident along the eyelid margins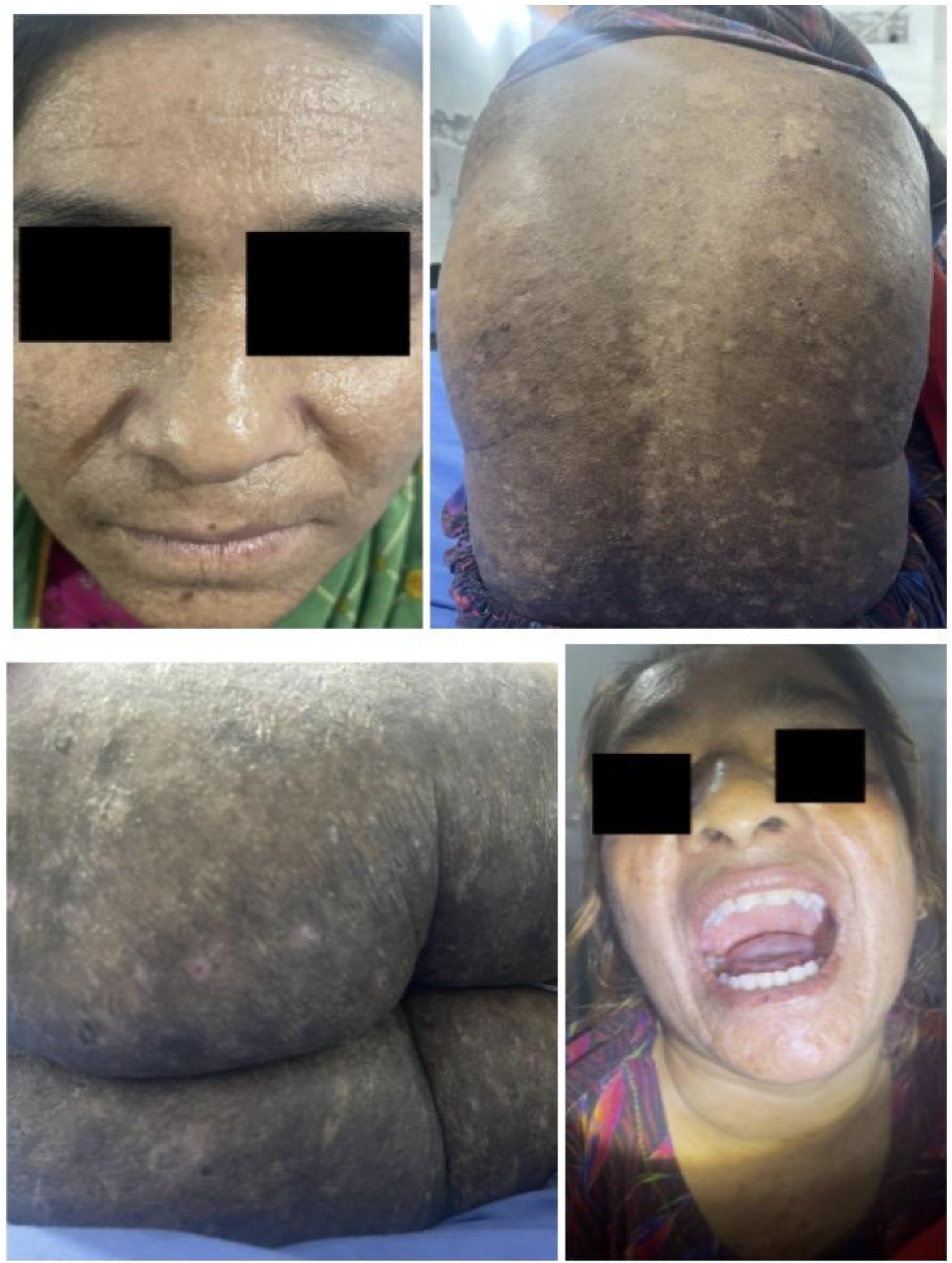
3.1.2. Case 2
A. Patient Information
A 16-year-old male presented with similar but more severe manifestations. B. Clinical Findings
Hoarseness of voice was noted at 2 months of age, followed by recurrent vesicles and papules that spread from the chest to involve the entire body within weeks. These lesions progressed to hyperkeratotic plaques with crusting and erosion.
Examination revealed widespread lesions over the back, abdomen, buttocks, and knees. Facial findings included pock-like scarring, accentuated perioral and periorbital lines, and a leonine-like facies. Moniliform blepharosis was prominent.
Oral mucosa showed yellowish infiltration with a markedly thickened frenulum limiting tongue protrusion. Associated symptoms included pruritus, photosensitivity, and drowsiness. Representative findings are shown in Figure 2. Clinical findings in Case 2 (Sibling 2); representative images of the 16-year-old male showing widespread vesiculopapular and hyperkeratotic plaque lesions with crusting and erosion, predominantly over the trunk and extensor surfaces. Facial features include pock-like scarring, accentuated periorbital and perioral lines, and a leonine-like appearance. Moniliform blepharosis is also present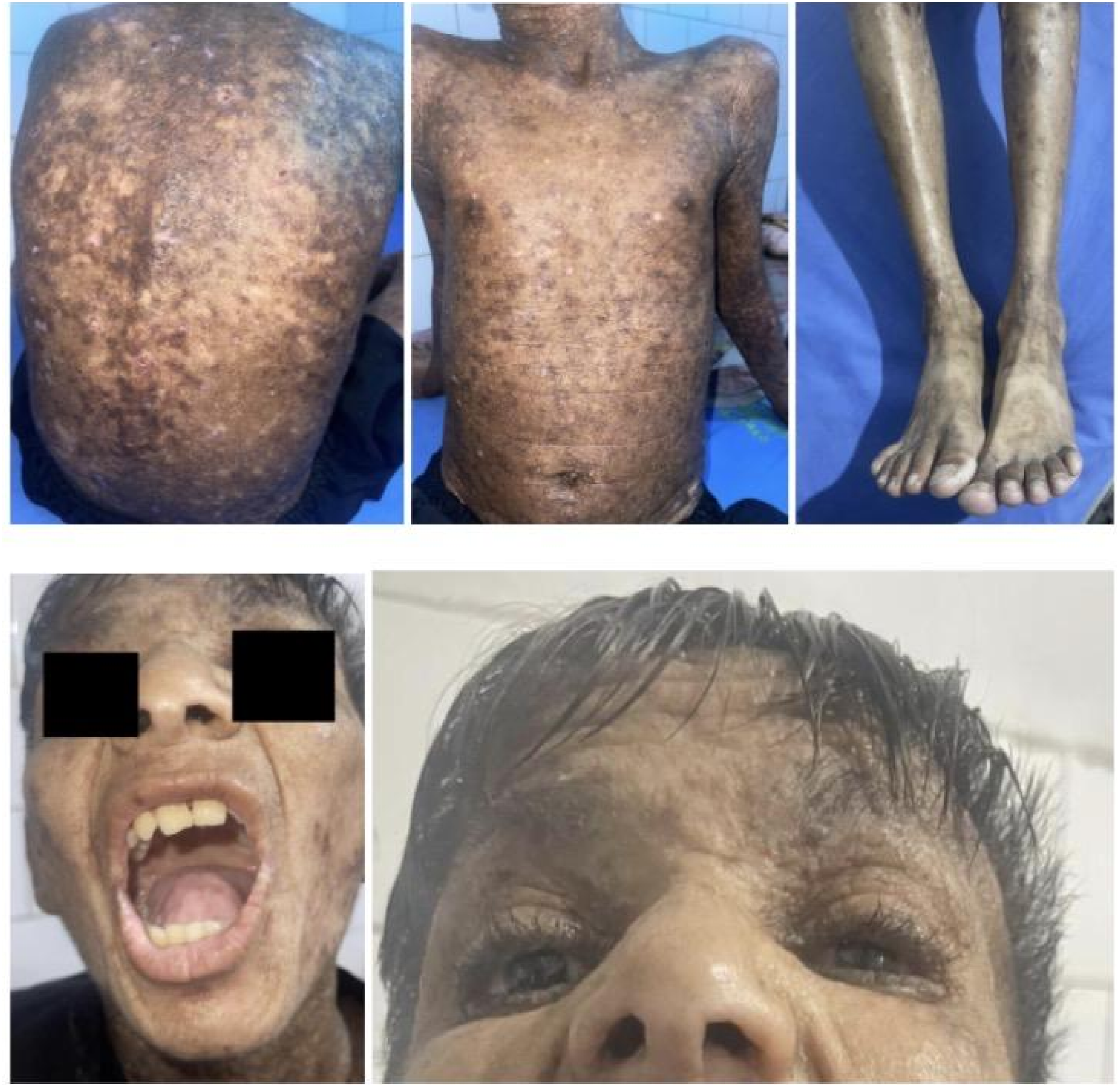
3.1.3. Case 3
A. Patient Information
A 25-year-old female presented with a clinical course similar to her siblings. B. Clinical Findings
The patient developed blistering lesions at approximately 2 months of age that healed with scarring and progressed to waxy papules and plaques. Lesions were predominantly located over extensor surfaces and the trunk, with progressive hyperpigmentation and hyperkeratosis.
Cutaneous findings included pock-like facial scarring, exaggerated perioral lines, and warty plaques over elbows and knees. Moniliform blepharosis was again evident.
Oral examination revealed mucosal infiltration and a thickened lingual frenulum restricting tongue movement. Persistent hoarseness of voice was present. Neurological symptoms included insomnia and irritability. Representative findings are shown in Figure 3. Clinical findings in Case 3 (Sibling 3); representative clinical images of the 25-year-old female demonstrating waxy papules and hyperkeratotic plaques, particularly over extensor surfaces, with associated facial scarring and exaggerated perioral lines. Moniliform blepharosis and features of mucocutaneous involvement are evident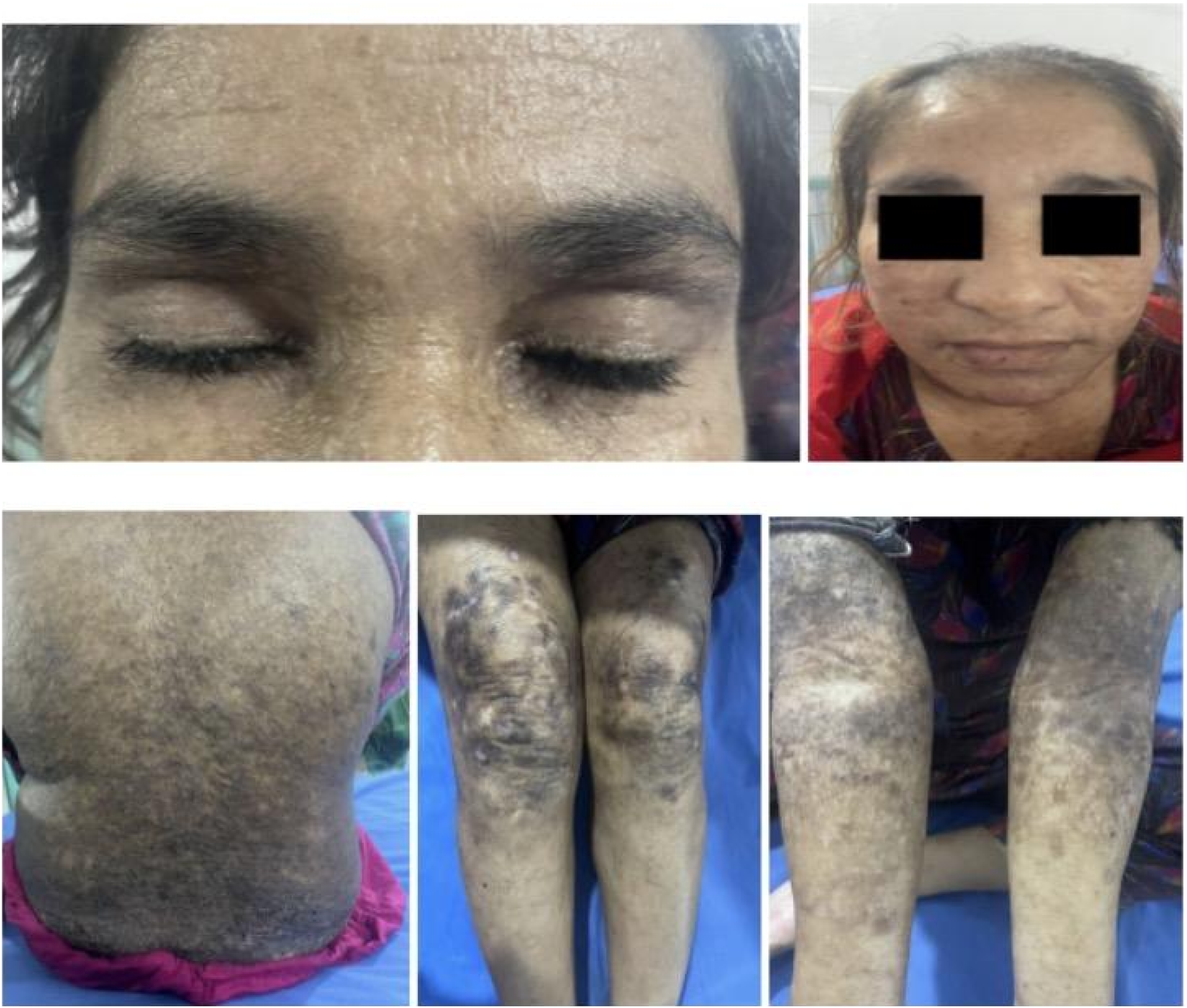
3.1.4. Comparative Clinical Summary
Comparative Clinical Features Across the Three Siblings With Lipoid Proteinosis
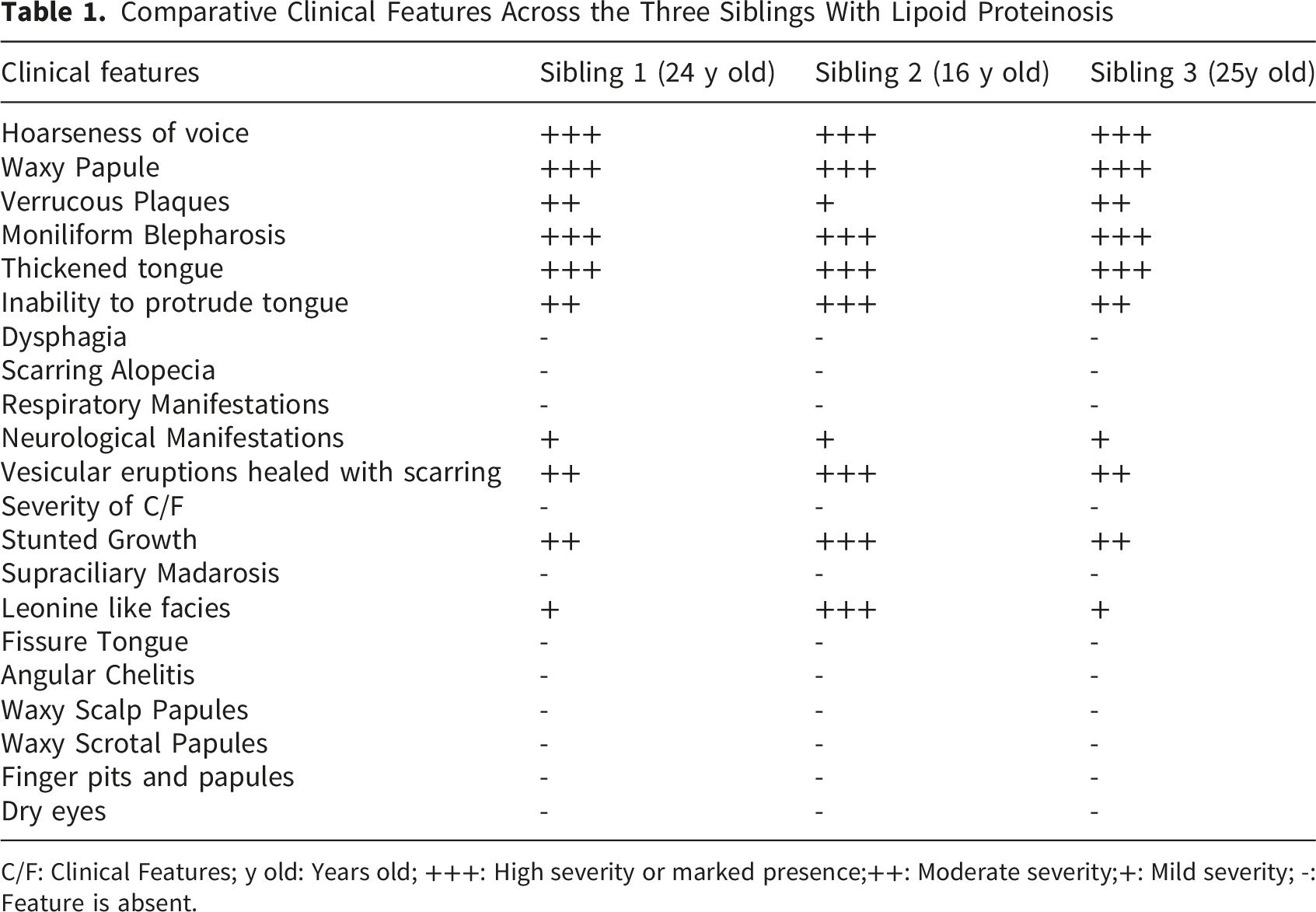
C/F: Clinical Features; y old: Years old; +++: High severity or marked presence;++: Moderate severity;+: Mild severity; -: Feature is absent.
3.2. Diagnostic Assessment
The diagnosis of lipoid proteinosis was established clinically based on the presence of characteristic features across all three siblings, including early-onset hoarseness of voice, recurrent vesiculopapular eruptions healing with scarring, moniliform blepharosis, and mucosal infiltration with restricted tongue movement.
Differential Diagnoses Considered and Distinguishing Features
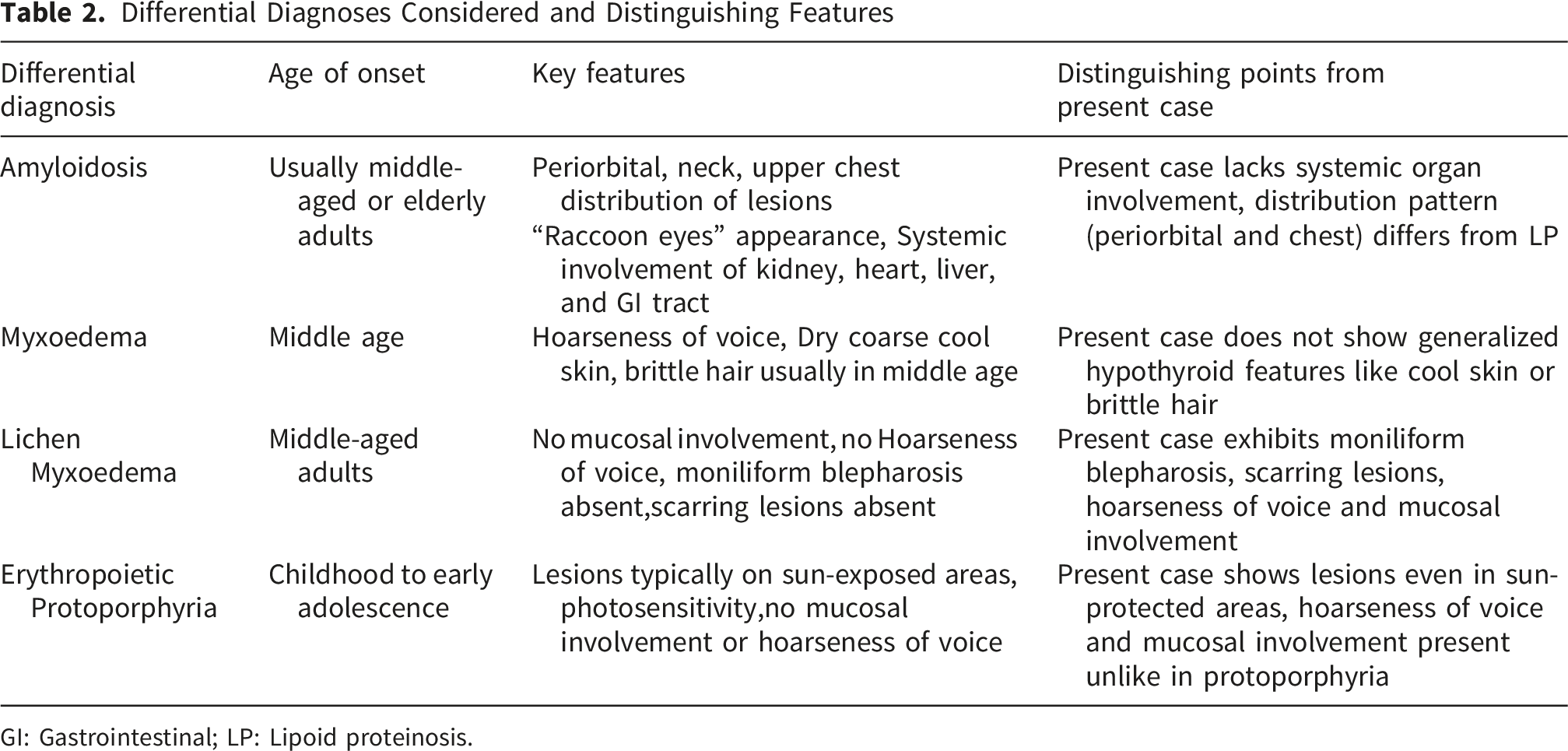
GI: Gastrointestinal; LP: Lipoid proteinosis.
Due to resource limitations, confirmatory investigations such as skin biopsy for PAS-positive hyaline deposition and ECM1 gene mutation analysis were not performed. Neuroimaging to detect intracranial calcifications was also not available. Despite these limitations, the classical clinical presentation provided strong diagnostic confidence.
3.3. Differential Diagnosis
Conditions considered included systemic amyloidosis, myxedema, lichen myxedematosus, and erythropoietic protoporphyria. These were excluded based on differences in clinical presentation, lesion distribution, age of onset, and lack of systemic or mucosal involvement. A detailed comparison is presented in Table 2.
3.4. Therapeutic Intervention
All three patients received similar management focused on symptomatic control. Baseline laboratory investigations were performed prior to initiating systemic therapy.
Oral acitretin was initiated at a dose of approximately 0.5 mg/kg/day to address hyperkeratotic lesions. Antihistamines, including cetirizine and fexofenadine, were prescribed for pruritus.
Topical therapy included mupirocin for eroded lesions and a 10% urea-based moisturizer to improve skin hydration. Patients were also advised to use emollients such as liquid paraffin and white soft paraffin regularly.
Supportive measures included counseling on trauma avoidance, sun protection, and general skin care.
3.5. Follow-Up and Outcomes
All three patients were followed for a period of approximately 12 weeks after initiation of therapy, with periodic outpatient evaluations at 4-week intervals. During this period, clinical monitoring focused on cutaneous response, pruritus severity, mucosal symptoms, and potential adverse effects of systemic retinoid therapy.
At the first follow-up (4 weeks), all patients reported a reduction in pruritus and decreased frequency of new vesiculobullous eruptions. Cutaneous examination demonstrated mild softening of hyperkeratotic plaques and reduced erythema in previously inflamed areas. However, established plaques and pock-like scars remained largely unchanged.
By the second follow-up (8 weeks), there was further stabilization of disease activity, with no significant progression of lesions. The hyperkeratotic plaques showed partial flattening, particularly over the trunk and flexural areas, although lesions over extensor surfaces such as elbows and knees remained more resistant to therapy. Photosensitivity symptoms improved modestly with adherence to photoprotective measures.
At 12 weeks, the overall response was characterized by symptomatic improvement rather than complete resolution. Pruritus was significantly reduced in all three patients, and no new widespread eruptions were observed. Hoarseness of voice persisted in all cases, with no appreciable improvement, reflecting the chronic structural involvement of the larynx.
Neurological and behavioral symptoms, including insomnia and irritability, showed minimal to no improvement, and were managed conservatively with supportive counseling. No acute neuropsychiatric deterioration was noted.
Importantly, no major adverse effects of acitretin therapy were observed during the follow-up period. Routine laboratory monitoring, including lipid profiles, remained within acceptable limits, and no patient required dose modification or discontinuation of therapy.
Despite partial clinical improvement, complete remission was not achieved, which is consistent with the known chronic and progressive nature of lipoid proteinosis. The patients were counseled regarding the need for long-term follow-up, adherence to treatment, and monitoring for potential complications, particularly airway involvement and progression of mucosal disease.
Further follow-up was planned; however, long-term outcomes could not be fully assessed at the time of reporting due to limitations in patient retention and resource constraints.
3.6. Timeline
Integrated Timeline of Clinical Course, Management, and Follow-Up Across all Three Cases
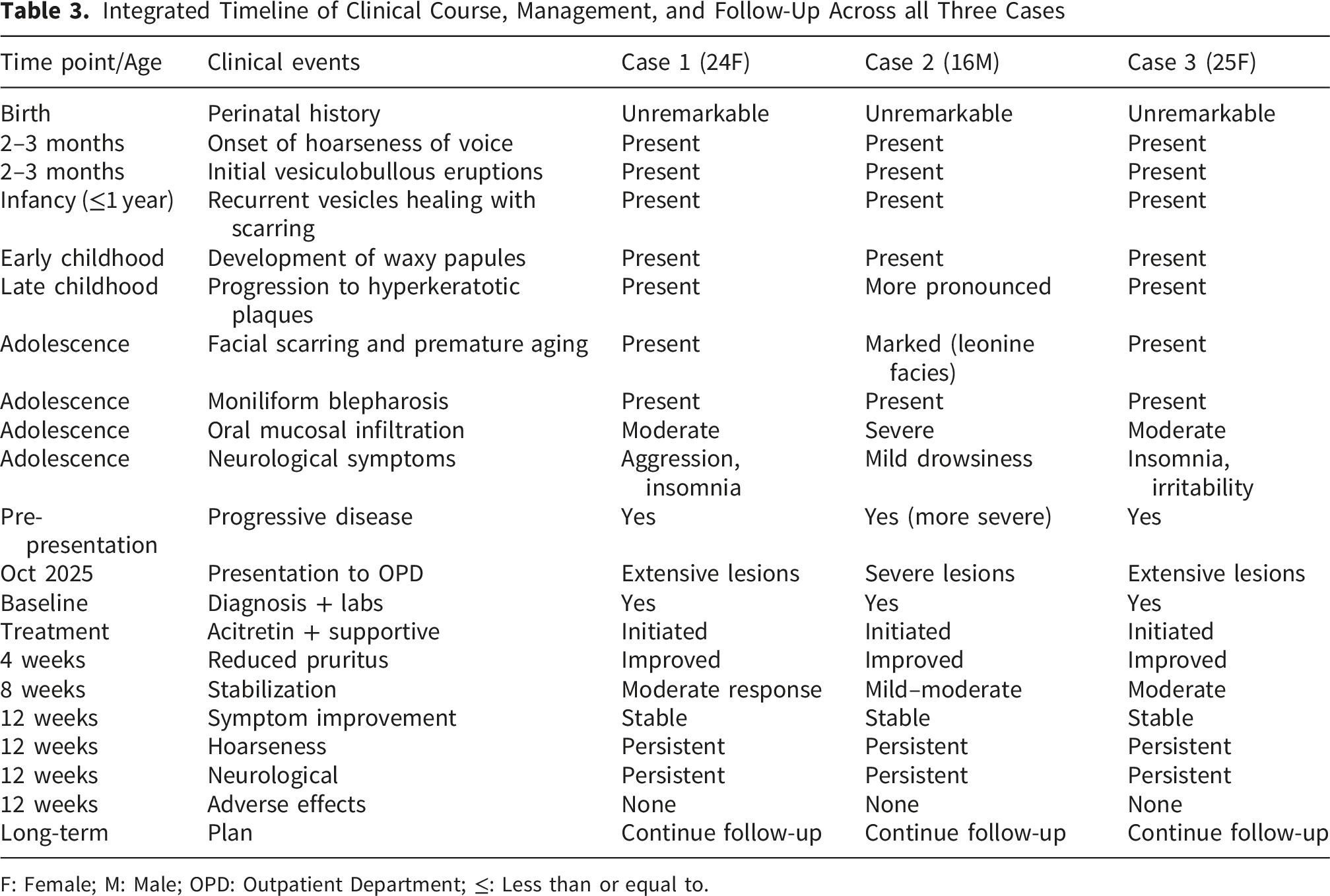
F: Female; M: Male; OPD: Outpatient Department; ≤: Less than or equal to.
3.7. Patient Perspective
The patients and their caregivers expressed significant psychosocial distress due to the chronic nature of the disease, visible skin lesions, and facial scarring. Hoarseness of voice further contributed to social discomfort. The family reported relief upon receiving a unifying diagnosis after years of uncertainty and expressed satisfaction with partial symptomatic improvement following treatment.
4. Discussion
Lipoid proteinosis (LP) is caused by loss of function variants in ECM1, leading to progressive deposition of hyaline-like material in the skin, mucosae, and, variably the central nervous system. 8 Clinically, onset of disease is typically in infancy with early breathy hoarseness, followed by waxy papules and vesicles that heal with pock-like scars and the pathognomonic moniliform blepharosis (beaded eyelid papules). 9 Oropharyngeal involvement most notably a thick, tethered lingual frenulum and tongue infiltration limits tongue protrusion and contributes to dysphonia. These features reflect impaired dermal scaffolding due to ECM1 deficiency, characterized histologically by thickened basement membranes and PAS-positive hyaline deposits around adnexal structures, vessels, and within mucosae. 10 Mechanistically, ECM1 is a secreted glycoprotein that binds and cross-links several basement-membrane and dermal matrix components—including perlecan, type IV collagen, laminin 332, and fibulins—and helps regulate matrix turnover, in part by inhibiting matrix metalloproteinase-9. 11 Loss of functional ECM1 therefore disrupts the orderly assembly of the basement membrane and dermal matrix, producing reduplication of basal laminae and progressive accumulation of the amorphous, PAS-positive hyaline (predominantly glycoprotein) material that defines the disease. 12 The anatomical site of deposition dictates the resulting phenotype: in the skin it produces waxy papules, hyperkeratotic plaques, and trauma-induced scarring, and along the eyelid margins the beaded papules of moniliform blepharosis; in the oral mucosa and lingual frenulum it causes infiltration and tethering that restrict tongue movement; in the larynx and vocal folds it causes the infiltration responsible for infant-onset hoarseness; and in the limbic system, particularly the amygdalae and adjacent temporal lobes, perivascular hyaline deposition and dystrophic calcification are thought to underlie the neuropsychiatric features such as anxiety, aggression, and disturbed sleep, which may be present even when calcification is not demonstrable on imaging.13,14 This structural fragility explains trauma-induced vesiculation and scarring, eyelid beading, and infant-onset hoarseness; photosensitivity and pruritus likely emerge when inflammatory triggers are superimposed on the underlying architectural defect. 15
This case series of three affected siblings illustrates the full phenotype and adds several points of clinical value. All three patients developed disease within 2–3 months of early life, then evolved the typical cutaneous and mucosal picture. Three of seven siblings being affected strongly supports autosomal-recessive inheritance and the absence of reported parental consanguinity highlights that LP can cluster in outbred families. 16 Every sibling demonstrated moniliform blepharosis, recurrent vesiculopapules coalescing into hyperkeratotic plaques (knees, elbows, buttocks, back), pock-like facial scarring with accentuated perioral/periorbital lines, and a short, thick frenulum with tongue infiltration. Together with persistent hoarseness, these features remain the strongest clinical signals for LP even when advanced tests are unavailable. Despite shared genetics, severity and emphasis differed (e.g., leonine-like facies, nail-fold atrophy, variable pruritus/photosensitivity),this mirrors prior observations that ECM1-related disease shows wide expressivity. 12 Aggression, insomnia, migraine, and drowsiness were reported. Neuropsychiatric features in LP do not require visible calcification and the absence of calcification on CT especially in young adults does not exclude LP.13,14
Based on the convergence of classic clinical findings across all three siblings, a clinical diagnosis of LP was made. Systemic amyloidosis typically shows a periorbital/neck/upper-chest distribution with “raccoon-eye” purpura and multisystem involvement—features absent in our case; instead, infancy onset with moniliform eyelid beading favored LP. 17 Myxedema (hypothyroidism) and lichen myxedematosus usually present in middle age with hypothyroid facies and, for lichen myxedematosus, no mucosal disease or blepharosis; our patients had early-life onset with mucosal involvement and eyelid beads. Erythropoietic protoporphyria produces photosensitive plaques confined to sun-exposed skin with mucosal sparing; hoarseness and eyelid beading are not expected. Our patients had mucosal disease and lesions even in protected areas. These contrasts, summarized in Table 2, support LP when histopathology or genetic confirmation is delayed.
There is no disease-modifying therapy; management is totally symptom-directed. All three patients followed the same treatmment. Baseline lipid profiles were obtained before initiating retinoids, which were prescribed at 0.5 mg/kg/day. Adjunct topical care included Polyfax ointment, mupirocin, and a 10% urea moisturizer to improve hydration and barrier function. Oral therapy comprised Acretrin 25 mg daily, cetirizine, and Estar, with fexofenadine added as needed for symptomatic relief. For maintenance, the 10% urea moisturizer and Acretrin 25 mg were continued, and patients were advised to apply an emollient mixture of liquid paraffin and white soft paraffin to reduce xerosis and scaling. Additional supportive options reported in the literature include emollients/antipruritics; cautious systemic retinoids for hyperkeratotic plaques; procedure-based treatments for focal disease (CO2/erbium laser, dermabrasion, blepharoplasty); frenulotomy for tongue tethering; and voice/airway care. Counseling on trauma avoidance, sun protection, and mental-health support is recommended.18-20
5. Limitations and Strengths
This series is notable for three infant-onset LP cases in a non-consanguineous Pakistani family, showing that autosomal recessive disorders can cluster outside consanguinity. Across siblings we captured the full diagnostic triad, moniliform blepharosis, hyperkeratotic scarring plaques, and oral tethering with hoarseness plus less-reported nail changes and clear intrafamilial heterogeneity. This case series has several limitations. First, the diagnosis was primarily clinical, and we could not provide histopathologic confirmation (e.g., PAS-positive hyaline deposits) or molecular confirmation (ECM1 sequencing), which limits definitive genotypic phenotype interpretation and may reduce diagnostic certainty in settings where phenotypic mimics exist. Second, neuroimaging was not performed (CT/MRI), so we could not evaluate for characteristic limbic calcifications or structural correlates of the reported neuropsychiatric symptoms, and subclinical CNS involvement may have been missed. Third, laryngeal assessment was limited by resource constraints, and lack of flexible fiber-optic laryngoscopy may have underestimated subtle vocal-cord or supraglottic infiltration contributing to hoarseness. Fourth, follow-up duration and standardized outcome measures were limited, preventing robust assessment of treatment response and adverse effects monitoring over time.
Our report, from a resource-constrained region of Pakistan where access to dermatopathology, genetic sequencing, and advanced imaging is limited, highlights how recognizing classic clinical patterns can sure the diagnosis. The convergence of cardinal signs across three siblings makes the diagnosis highly credible. Where feasible, next steps include ECM1 sequencing; dermatopathology to demonstrate PAS-positive deposits and targeted MRI of limbic structures if neuropsychiatric symptoms emerge or progress. Following variant identification, family screening and premarital/antenatal counseling are advisable.21,22 With this case series, we underscore that Urbach–Wiethe disease typically begins in childhood and is marked by early hoarseness and characteristic skin findings—features that can enable early detection and timely, symptom-directed management even in low-resource settings.
6. Conclusion
In conclusion, this case series describes three siblings from a non-consanguineous Pakistani family, illustrating the full diagnostic triad of lipoid proteinosis: moniliform blepharosis, hyperkeratotic scarring plaques, and oral tethering with persistent hoarseness. Despite the lack of access to advanced dermatopathology, genetic sequencing, or fiber-optic laryngoscopy, the convergence of these cardinal signs across multiple family members allows for a highly credible clinical diagnosis. While complete remission was not achieved, symptomatic management with oral acitretin and topical therapies provided meaningful relief from pruritus and stabilized new lesion formation. These findings underscore that early recognition of classic clinical patterns can facilitate timely, symptom-directed management and essential genetic counseling even in resource-limited settings.
7. Informed Consent
Written informed consent was obtained from the adult patients and the legal guardian of the minor patient for the publication of this case series and any accompanying images. The participants were informed that their identity would be protected through the use of de-identified clinical data and the masking of eyes in photographs to ensure anonymity. A copy of the written consent is availabsle for review upon request.
Footnotes
Ethical Considerations
Our institution does not require formal ethical approval for reporting individual case series.
Consent to Participate
The patients and the legal guardian of the minor patient provided written informed consent to participate in this study and for the use of their anonymized information.
Consent for Publication
Written informed consent was obtained from the adult patients and the legal guardian of the minor sibling for the publication of this case series and all accompanying clinical images.
Author Contributions
M.M., I.Z., B.M., A.S., and A.A.R. were involved in the conception and design of the study. M.M., I.Z., B.M., A.S., and A.A.R. contributed to data acquisition, case identification, and clinical management of patients. M.M., I.Z., B.M., A.S., A.A.R., and A.Sa. (Abedin Samadi) contributed to data analysis and interpretation. M.M. and I.Z. drafted the initial manuscript. B.M., A.S., A.A.R., and A.Sa. critically revised the manuscript for important intellectual content. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work.
Funding
This study was not funded by any entity and was conducted in a resource-limited setting.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data supporting the findings of this case series are available from the corresponding author upon reasonable request. Due to the nature of the study, patient confidentiality and ethical considerations regarding rare genetic disorders prevent the public sharing of sensitive clinical data. All other data related to the study, including de-identified information and clinical summaries, can be made available upon request in accordance with the journal’s data-sharing policies.