
Abstract
Background
Congenital anomalies of the urogenital system frequently coexist with renal abnormalities due to their shared origin from the intermediate mesoderm. Skeletal anomalies may also be associated, reflecting overlapping embryological development of mesodermal derivatives. Such multisystem involvement can pose a diagnostic challenge, particularly when identified incidentally.
Case Presentation
We report a 15-year-old female who presented with abdominal swelling and pain, later diagnosed as an umbilical hernia. During radiological evaluation, incidental findings of right renal agenesis, scoliosis, and a bicornuate uterus were identified. The patient underwent successful surgical management of the umbilical hernia. This case highlights the importance of recognizing coexisting anomalies and adopting an integrated approach in evaluation and management.
Conclusion
This case highlights the need for a systematic, bidirectional evaluation when a congenital anomaly is identified, particularly involving the renal, genital, and skeletal systems. A syndromic perspective is essential to avoid missed diagnoses, facilitate appropriate follow-up, and anticipate long-term implications in such patients.
Keywords
Introduction
Congenital anomalies of the urogenital tract are often accompanied by abnormalities in other systems, notably the renal and skeletal systems. Müllerian duct anomalies have been reported to coexist with renal defects in approximately 30–40% of cases.1,2 Well recognized associations include a unicornuate uterus with ipsilateral renal agenesis and conditions such as Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome, which may present with vertebral anomalies. 3
The urinary and genital systems share an embryological origin in the intermediate mesoderm, which may explain the frequent association of Müllerian anomalies with renal abnormalities.4,5 On the other hand, skeletal abnormalities such as scoliosis arise from disruptions in the development of somites, which are derived from the paraxial mesoderm. Somites differentiate into the sclerotome, dermatome, and myotome, with the sclerotome contributing to vertebral formation through processes of segmentation and resegmentation. Disturbances in this process can lead to vertebral anomalies, including scoliosis.
The coexistence of skeletal, renal, and Müllerian anomalies can be explained by their origin from adjacent mesodermal structures and their overlapping periods of development, particularly between the third and eighth weeks of gestation. Such temporal and spatial proximity increases the likelihood of concurrent maldevelopment, resulting in multisystem anomalies. 6 Therefore, the identification of a single congenital anomaly should prompt a systematic evaluation for associated defects, 7 especially in structures sharing common embryological origins.
In this case report, we present a rare coexistence of multisystem anomalies involving the skeletal, renal, and urogenital systems in an adolescent female.
Case Discussion
A 15-year-old female presented to the outpatient department with complaints of intermittent diffuse abdominal pain for the past 2 months. The pain was aggravated by food intake and relieved on lying down. She also reported a history of abdominal swelling since childhood. Additionally, she complained of constipation. There was no history of trauma, fever, weight loss, or vomiting.
She attained menarche at 13 years of age. Her menstrual history is significant for regular cycles associated with heavy menstrual bleeding and prolonged duration of flow. The patient was born to a non-consanguineous marriage, with no significant family history of congenital abnormalities or other relevant comorbidities. On examination, a single oval-shaped swelling measuring approximately 3 × 3 cm was noted in the umbilical region, with no overlying skin changes. On palpation, the swelling was soft in consistency and completely reducible; no cough impulse was elicited. A fascial defect measuring approximately 2 × 2 cm was palpable at the umbilicus. Spinal asymmetry was noted, with lateral deviation of the vertebral column. Per rectal examination revealed an external hemorrhoid.
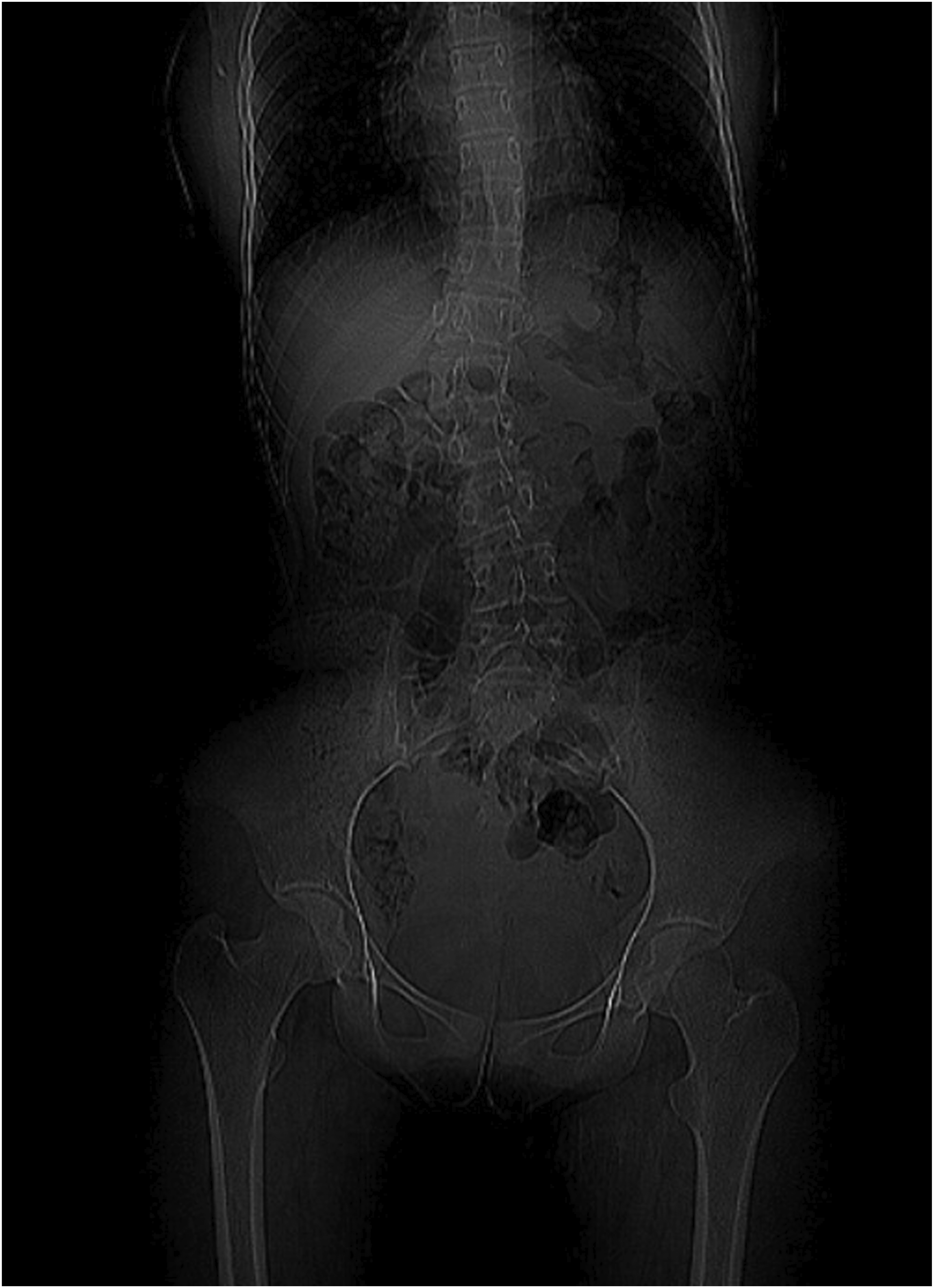
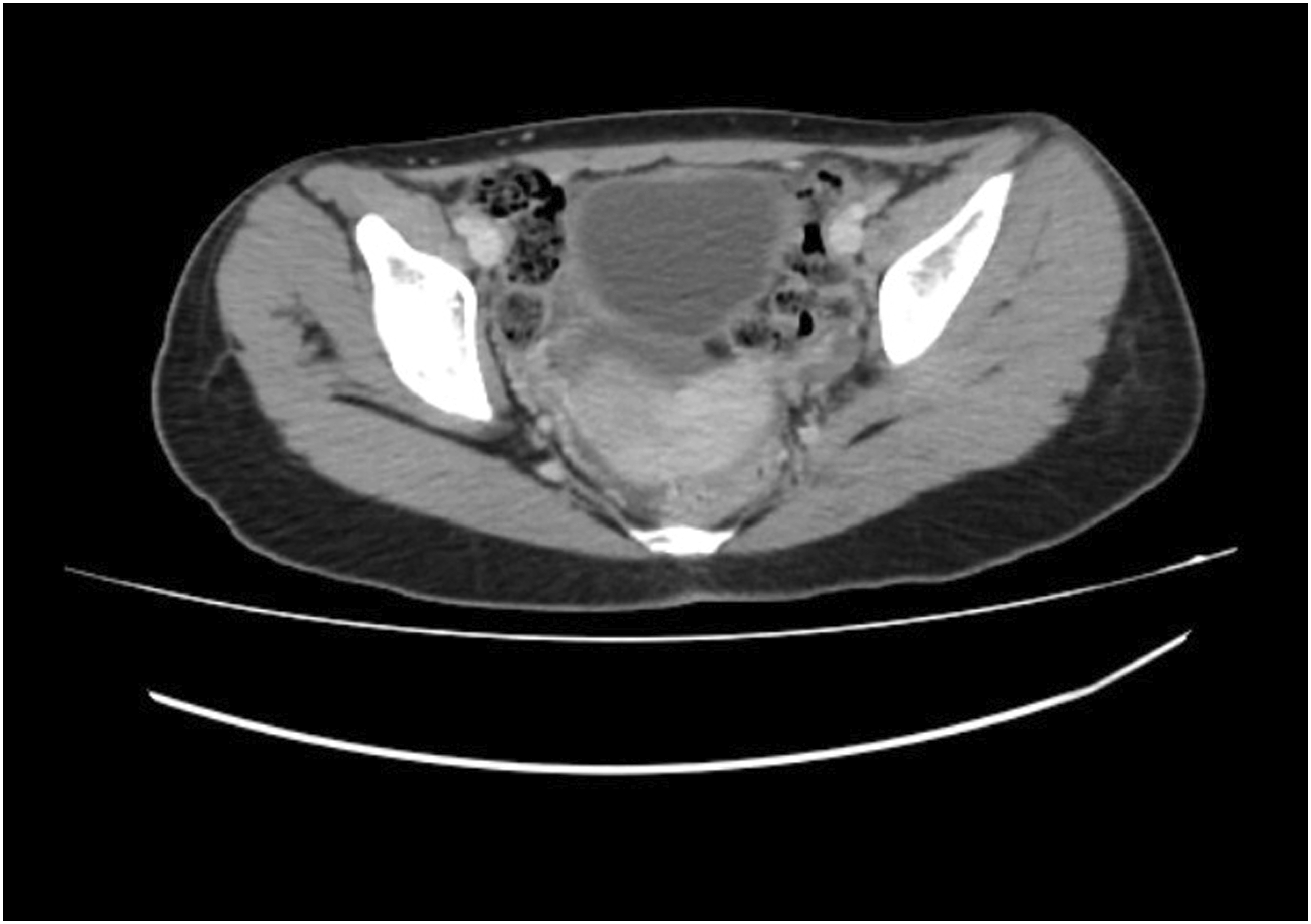
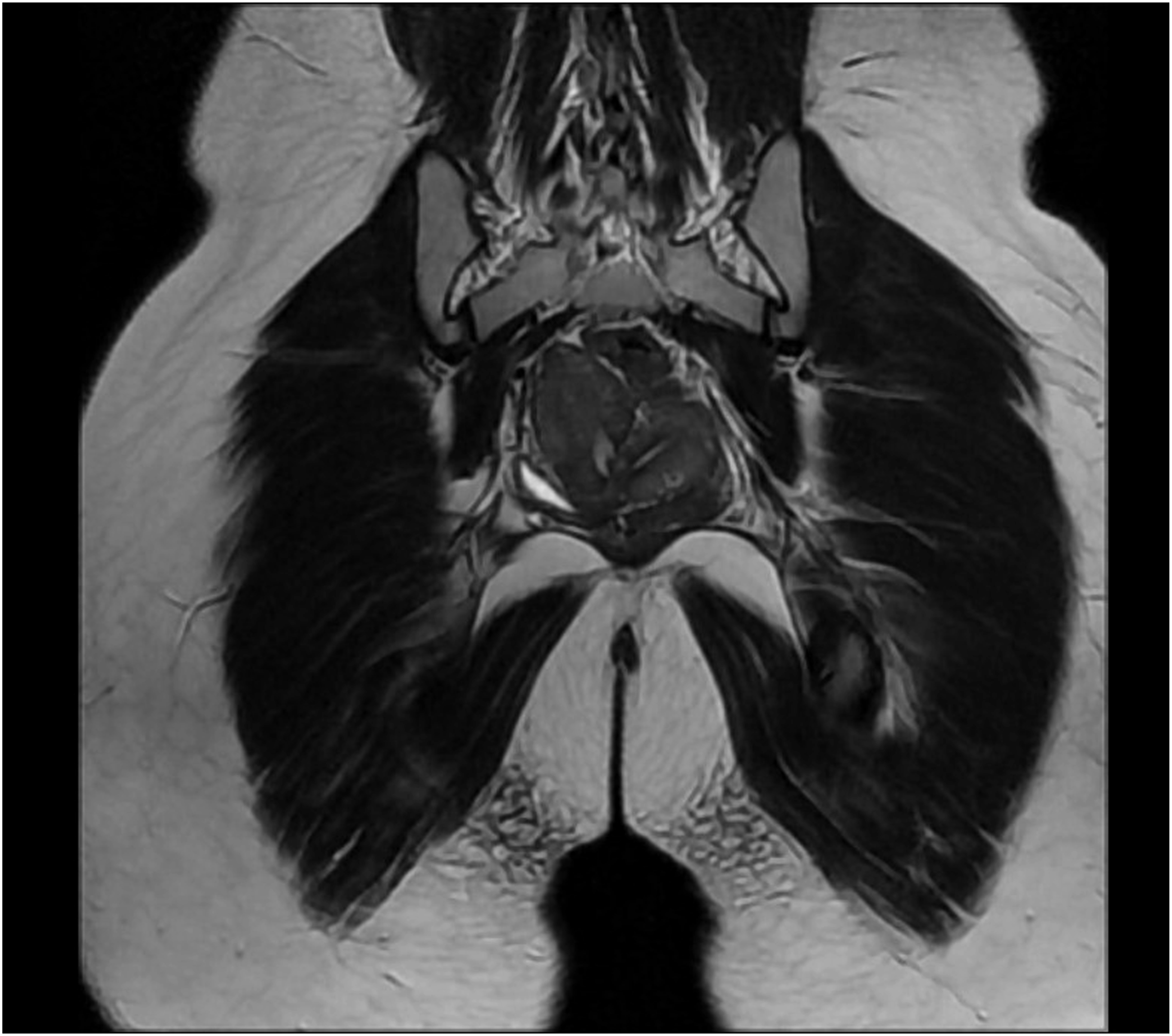
Ultrasound of the abdomen revealed a 4 mm defect in the anterior abdominal wall with herniation of omental fat, suggestive of an umbilical hernia. The right kidney was not visualized in the right renal fossa. X-ray of the spine demonstrated a right-sided lumbar scoliosis with a Cobb angle of approximately 20°. These findings were further confirmed on contrast-enhanced CT (CECT) of the abdomen, which showed the absence of the right kidney, consistent with right renal agenesis (Figure 1) and scoliosis (Figure 2). The left kidney was enlarged, suggestive of compensatory hypertrophy. The umbilical hernia with a corresponding wall defect was also visualized. CT imaging demonstrated a subtle indentation in the uterine contour, imparting a heart-shaped appearance and raising suspicion for a Müllerian anomaly. This finding was consistently demonstrated on MRI, which confirmed the diagnosis of a bicornuate uterus (Figures 3 and 4). Genetic testing was not performed due to resource limitations and limited immediate impact on clinical management, as the diagnosis and treatment plan were guided primarily by the patient’s clinical presentation and radiological findings. Contrast-enhanced CT abdomen (coronal section) demonstrating absence of the right kidney in the right renal fossa, consistent with right renal agenesis CT topogram (scout view) demonstrating right-sided lumbar scoliosis of the spine Contrast-enhanced CT abdomen (axial section) demonstrating an indentation in the uterine cavity MRI pelvis (coronal section) demonstrating two uterine cavities separated by a fundal cleft, suggestive of a possible bicornuate uterus.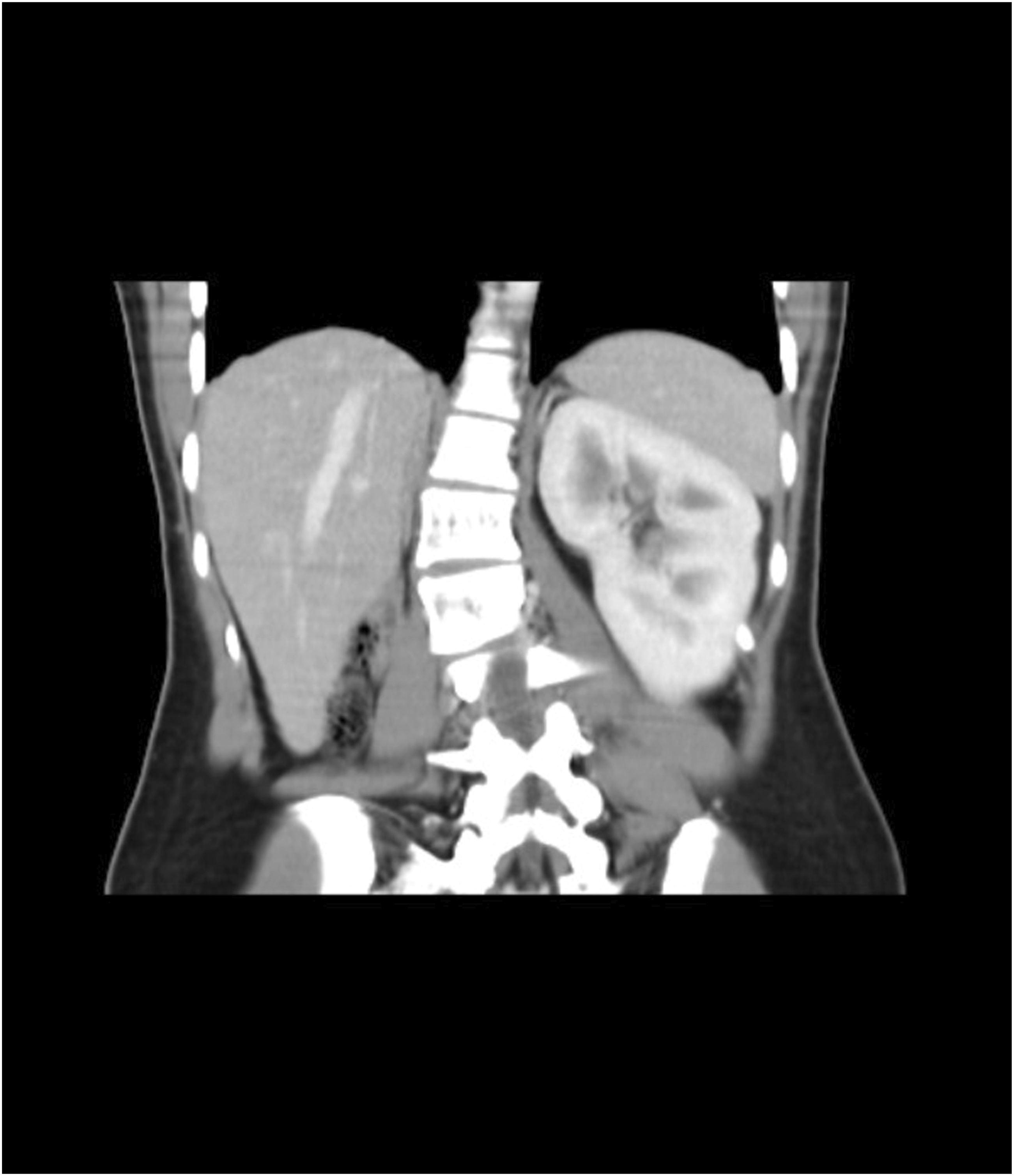
In conclusion, the patient was diagnosed with an umbilical hernia, along with incidental findings of right renal agenesis, right-sided lumbar scoliosis, and a bicornuate uterus. She underwent successful surgical management of the umbilical hernia with preperitoneal mesh repair and had an uneventful postoperative recovery. A multidisciplinary approach with appropriate follow-up was advised for the associated congenital abnormalities to ensure long-term monitoring and management.
Discussion
The umbilical opening, a defect in the linea alba derived from embryonic mesoderm, normally closes by obliteration of the umbilical ring, influenced by local mechanical factors and rectus abdominis muscle tone. Failure of complete closure of the umbilical ring allows protrusion of intra-abdominal contents, resulting in an umbilical hernia. 8 Umbilical hernia has been reported in association with several congenital and genetic conditions, including Beckwith-Wiedemann syndrome, Down syndrome, and Congenital hypothyroidism. It may also be observed in connective tissue disorders such as Ehlers-Danlos syndrome and Marfan syndrome, as well as in Mucopolysaccharidoses. Although the direct association between umbilical hernia and genitourinary anomalies remains controversial, a common mesodermal origin or an underlying defect in connective tissue formation may explain their coexistence in this patient.
Renal agenesis results from failure of interaction between the ureteric bud and the metanephric mesenchyme during embryogenesis. 9 Given the close developmental relationship between the urinary and genital systems, urinary tract anomalies are frequently associated with genital abnormalities, as seen in conditions such as Mayer-Rokitansky-Küster-Hauser syndrome, Herlyn-Werner-Wunderlich syndrome, Fraser syndrome, and Kallmann syndrome. 10 Individuals with a solitary functioning kidney are often asymptomatic due to compensatory hypertrophy of the contralateral kidney. However, in the context of congenital anomalies of the kidney and urinary tract, a solitary functioning kidney may be associated with additional urinary tract abnormalities such as vesicoureteral reflux. In the present case, no such associated urinary tract abnormalities were identified. 11
A bicornuate uterus results from incomplete fusion of the paired Müllerian ducts during the 6th–10th weeks of embryogenesis, leading to two endometrial cavities with a single or double cervix. This anomaly arises from disruption of the ductal fusion and differentiation process, resulting in partial separation of the uterine horns. Adolescents are often asymptomatic, although some may present with menstrual irregularities such as menorrhagia or dysmenorrhea, as seen in this patient. Physical examination findings are typically unremarkable. 12
Scoliosis may arise from genetic and molecular disturbances that impair normal vertebral formation and segmentation during somite development, typically between the third and fifth weeks of gestation. 13 It has been reported in association with syndromic conditions such as Ehlers–Danlos syndrome, osteogenesis imperfecta, Marfan syndrome, and DiGeorge syndrome. It may also be seen in association with Müllerian anomalies, as observed in this case. 14
A plausible embryologic hypothesis is that this adolescent has a developmental field defect affecting the Müllerian system, urinary tract, and axial skeleton rather than an isolated disease. Although the pattern of anomalies resembles the MRKH syndrome, the presence of normal menstruation makes the diagnosis unlikely.15,16
This case is similar in organ clustering and possible embryologic timing to the MURCS association, since MURCS is defined by the Müllerian duct and renal anomalies, along with cervicothoracic vertebral dysplasia. However, MURCS typically presents with primary amenorrhea, making her phenotype closer to a partial Mullerian anomaly with associated renal and skeletal maldevelopment rather than full MURCS.
A suitable explanation would be a disruption around the fourth week of embryogenesis, when mesodermal patterning and spatial relationships between pronephric structures, Müllerian precursors, and cervicothoracic somites are established. Although the Müllerian ducts develop later, an early insult at this stage may disrupt this developmental field, resulting in a localized mesodermal defect and the cluster of anomalies observed in this case.
VACTERL association is one of the differential diagnoses considered for this case. It is the presence of at least three of the following congenital malformations: vertebral defects, anal atresia, cardiac defects, tracheo-oesophageal fistula, renal anomalies, and limb abnormalities. In addition to these core component features, patients may also have other congenital anomalies. 17 Since this patient has only two, that is, a vertebral defect (scoliosis) and a renal anomaly (unilateral renal agenesis), of the congenital malformations, it was later not regarded.
Another differential considered is Herlyn-Werner-Wunderlich syndrome, also called as obstructed hemivagina and ipsilateral renal anomaly (OHVIRA). It is characterized by the association of a septate uterus, obstructed hemivagina, and ipsilateral renal agenesis. 18 Patients do have normal menstruation from the unobstructed side of the uterus/vagina. However, they usually present shortly after menarche with worsening dysmenorrhea or a palpable pelvic mass due to hematocolpos. 19 As she is asymptomatic and menstruating normally, a blind hemivagina is ruled out.
Klippel-Feil syndrome (KFS) is defined as the congenital fusion of two or more cervical vertebrae, resulting from a failure of segmentation during spinal development. 20 Our patient has scoliosis, which leans towards it, but does not exhibit clinical signs such as a short neck, low posterior hairline, and restricted neck mobility, thus eliminating the possibility of KFS.
The coexistence of renal and Müllerian anomalies in this case highlights the well-established embryological connection between the mesonephric and paramesonephric duct systems, necessitating a systematic evaluation when one anomaly is identified. Specifically, the detection of congenital renal anomalies, such as unilateral renal agenesis, should prompt targeted assessment of the reproductive tract, as up to 30-40% of patients with Müllerian duct anomalies may have associated urinary tract abnormalities.1,21 Conversely, identification of a uterine malformation (e.g., bicornuate uterus) might warrant renal imaging to exclude occult urological defects. 22 This screening approach can help to avoid missed diagnosis, particularly in resource-limited settings where anomalies may remain clinically silent. From a prognostic standpoint, Mullerian anomalies might be associated with significant reproductive morbidity, including increased risks of infertility, recurrent pregnancy loss, preterm delivery, malpresentation, and obstetric complications. 23 Additionally, unilateral renal agenesis, though often clinically asymptomatic, is associated with long-term risks such as hypertension, proteinuria, and chronic kidney disease due to hyperfiltration in the solitary kidney,24,25 while coexisting scoliosis may predispose to chronic pain, progressive deformity, and, in severe cases, restrictive pulmonary impairment. 26 Early recognition might enable anticipatory counseling, fertility planning, and high-risk obstetric surveillance, which may help meaningfully improve reproductive outcomes. Therefore, this case highlights the importance of adopting a syndromic perspective in congenital anomalies and emphasizes the need for integrated, multidisciplinary evaluation protocols when a single developmental abnormality is identified.
The finding of Scoliosis in this patient, which is incidental and not associated with any deformities, pain, or functional limitations, should be managed with observation and periodic orthopedic follow-ups. Long-term management focuses on preserving renal function and preventing complications associated with a solitary functioning kidney. This includes periodic monitoring of renal parameters, including serum creatinine, urinalysis, urinary albumin, and estimated glomerular filtration rate. Regular blood pressure assessment is essential for early detection and management of hypertension. Patients should also be counseled on avoiding nephrotoxic medications and on the importance of maintaining adequate hydration, with consideration of periodic nephrology follow-up as part of ongoing care. 27
Management of a bicornuate uterus depends largely on the patient’s clinical presentation and obstetric history. In asymptomatic individuals with normal menstruation, as in this case, no immediate intervention is required. However, patients should be counseled regarding potential reproductive complications, including recurrent pregnancy loss, preterm labor, and malpresentation. Surgical correction, such as metroplasty, is generally reserved for selected cases with a history of adverse obstetric outcomes, particularly recurrent pregnancy loss or preterm delivery attributable to the uterine anomaly. In future pregnancies, close antenatal surveillance with serial ultrasonography and cervical length monitoring is recommended to optimize outcomes. 12
The novelty of this case lies in the atypical coexistence of renal, Müllerian, skeletal, and abdominal wall anomalies in a menstruating adolescent without a definitive syndromic diagnosis. A potential pitfall in such cases is focusing primarily on the presenting complaint, which may result in associated congenital anomalies being overlooked. Failure to recognize this multisystem involvement may delay appropriate management of renal, reproductive, and musculoskeletal complications. Thus, detection of a congenital renal anomaly should prompt targeted pelvic imaging to evaluate for associated Müllerian abnormalities, while identification of a uterine anomaly should similarly warrant renal imaging and, when indicated, skeletal assessment. This bidirectional screening approach may facilitate earlier recognition of coexisting developmental anomalies. Such an approach can help with well-timed diagnosis and enable multidisciplinary management.
Conclusion
This case describes a rare combination of coexisting urogenital and skeletal anomalies, highlighting the importance of recognizing shared embryological pathways in congenital malformations. It also highlights the diagnostic challenge posed by such presentations, as clinically unrelated abnormalities may easily be overlooked unless a systematic evaluation is undertaken. Incidental findings should not be dismissed, as they may indicate an underlying developmental association or syndrome. The coexistence of renal, Müllerian, and skeletal defects reinforces the need for thorough assessment in young patients with congenital anomalies, particularly because some abnormalities may remain asymptomatic for years. Early detection of associated findings facilitates timely multidisciplinary follow-up, appropriate reproductive and prognostic counseling, and better anticipation of future complications. Overall, these observations support a systematic, syndromic, and diagnostically vigilant approach to evaluating congenital anomalies in clinical practice.
Limitations
This case has certain limitations. First, genetic evaluation, including chromosomal and molecular testing, could not be performed due to limited access to advanced diagnostic facilities, which limited our ability to identify any underlying genetic or chromosomal abnormality that may explain the observed coexisting anomalies. As a result, the diagnosis remains based primarily on clinical and radiological findings, and a definitive syndromic classification could not be established. Additionally, long-term follow-up may be challenging given the patient’s socioeconomic circumstances, which may limit ongoing surveillance and assessment of future renal, reproductive, and musculoskeletal outcomes.
Footnotes
Ethical Considerations
Written informed consent was obtained from the patient and/or legal guardian for publication of this case report and accompanying images.
Consent to Participate
Written informed consent for participation and publication was obtained from the patient’s parent/legal guardian.
Consent for Publication
Written informed consent was obtained from the patient to publish this report per the journal’s patient consent policy.
Author Contributions
DA was responsible for case identification, obtaining informed consent, and conceptualizing the structure of the case report. RG contributed to editing, critical revision, and final submission of the manuscript. MK, PC, SA, RD, SR, KC, PM, and RB were involved in drafting and writing the manuscript. All authors reviewed and approved the final version of the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.