
Abstract
Introduction
Congenital coronary artery anomalies are uncommon but clinically significant conditions associated with myocardial ischemia, malignant ventricular arrhythmias, and sudden cardiac death, particularly in young individuals. Among these anomalies, anomalous origin of the right coronary artery from the pulmonary artery (ARCAPA) is exceptionally rare, with a reported prevalence of approximately 0.002%. Although ARCAPA may remain clinically silent for many years due to collateral circulation from the left coronary system, it carries a persistent risk of myocardial ischemia caused by a coronary steal phenomenon and may result in sudden cardiac death.
Case Presentation
We report the case of a 19-year-old Somali male with no prior medical history who presented with a several-month history of recurrent syncope occurring both at rest and during exertion, without preceding chest pain, palpitations, dyspnea, seizure-like activity, or prodromal symptoms. He presented following a prolonged syncopal episode and was found to be hemodynamically unstable, with a blood pressure of 86/54 mmHg and sinus tachycardia. Initial laboratory investigations, including complete blood count, renal function, serum electrolytes, coagulation profile, and cardiac biomarkers, were within normal limits. Electrocardiography demonstrated diffuse ST-segment depression in the inferolateral leads with reciprocal ST-segment elevation in lead aVR, raising concern for global subendocardial ischemia. Transthoracic echocardiography revealed preserved left ventricular systolic function, with an ejection fraction of 65%, a mildly dilated main pulmonary artery, and mild pulmonary regurgitation. Due to the unavailability of coronary computed tomography angiography, urgent invasive coronary angiography was performed approximately 4 hours after presentation. Angiography revealed anomalous origin of the right coronary artery from the pulmonary artery with retrograde flow into the pulmonary trunk, consistent with a significant coronary steal phenomenon. No obstructive coronary artery disease was identified. Approximately 30 minutes after angiography, the patient developed sudden hemodynamic collapse and progressed to cardiac arrest. Despite prolonged advanced cardiopulmonary resuscitation, return of spontaneous circulation was not achieved. The presumed cause of death was malignant ventricular arrhythmia secondary to myocardial ischemia related to ARCAPA; however, the exact mechanism could not be definitively established.
Discussion
This case highlights the potential for rapid fatal deterioration in ARCAPA despite preserved ventricular systolic function. Recurrent syncope and ischemic electrocardiographic abnormalities should be recognized as high-risk features. The electrocardiographic pattern observed may reflect diffuse myocardial ischemia due to coronary steal rather than atherosclerotic coronary artery disease.
Conclusion
ARCAPA should be considered in young patients presenting with unexplained recurrent syncope and high-risk electrocardiographic findings. Early recognition, appropriate use of multimodality imaging, close monitoring, and prompt surgical referral are essential to prevent catastrophic outcomes.
Keywords
Introduction
Congenital anomalies of the coronary arteries are uncommon but clinically significant conditions that may cause myocardial ischemia, ventricular arrhythmias, heart failure, and sudden cardiac death, particularly in young individuals without traditional cardiovascular risk factors. 1 Among these anomalies, anomalous origin of the right coronary artery from the pulmonary artery (ARCAPA) is exceptionally rare, with an estimated prevalence of approximately 0.002% in the general population. It occurs less frequently than anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA).2,3
Unlike ALCAPA, which commonly presents in infancy with heart failure and left ventricular dysfunction, ARCAPA may remain clinically silent until adolescence or adulthood. This delayed presentation is usually explained by the development of collateral circulation from the left coronary artery to the right coronary system. However, the presence of collateral circulation does not eliminate risk. Instead, oxygenated blood may flow retrogradely from the left coronary system through the right coronary artery and drain into the low-pressure pulmonary artery. This produces a coronary steal phenomenon, reducing effective myocardial perfusion and predisposing patients to ischemia, exertional symptoms, syncope, ventricular arrhythmias, and sudden cardiac death.2-5
The clinical presentation of ARCAPA is highly variable. Some patients are diagnosed incidentally, while others present with angina, dyspnea, heart failure, myocardial infarction, arrhythmia, or sudden cardiac arrest.3,6,7 Syncope is an especially important symptom because it may reflect transient global myocardial ischemia or intermittent malignant ventricular arrhythmia. In young patients, recurrent unexplained syncope with ischemic electrocardiographic changes should therefore prompt urgent evaluation for structural and congenital coronary abnormalities.
Diagnosis of ARCAPA requires demonstration of the anomalous origin and abnormal coronary flow pattern. Coronary computed tomography angiography is useful for defining coronary anatomy noninvasively, while invasive coronary angiography can demonstrate retrograde filling of the right coronary artery and drainage into the pulmonary artery. Multimodality imaging, including echocardiography, coronary computed tomography angiography, cardiac magnetic resonance imaging, and invasive angiography, plays an important role in diagnosis, risk assessment, and surgical planning. 8
Surgical correction is generally recommended after diagnosis, even in asymptomatic patients, because of the persistent risk of ischemia and sudden cardiac death. Surgical strategies include direct reimplantation of the anomalous right coronary artery into the aorta, ligation of the anomalous origin with coronary artery bypass grafting, or other individualized repair techniques depending on anatomy and institutional expertise.7,8
This case is notable because it describes a young adult with recurrent syncope, diffuse subendocardial ischemic electrocardiographic changes, preserved ventricular systolic function, angiographic evidence of coronary steal, and sudden death before surgical correction. It emphasizes the narrow diagnostic and therapeutic window in symptomatic ARCAPA and highlights the need for urgent recognition and management.
Case Presentation
A 19-year-old Somali male with no known chronic medical illness presented to the emergency department after a prolonged episode of syncope. He reported a several-month history of recurrent syncopal episodes. The episodes occurred both at rest and during exertion, were sudden in onset, and were not preceded by chest pain, palpitations, dyspnea, dizziness, seizure-like activity, or other prodromal symptoms. There was no known family history of sudden cardiac death, congenital heart disease, cardiomyopathy, or inherited arrhythmia syndrome.
On arrival to the emergency department, the patient was conscious but hemodynamically unstable. His blood pressure was 86/54 mmHg, heart rate was 118 beats/min, respiratory rate was 22 breaths/min, oxygen saturation was 97% on room air, and body temperature was 36.8°C. Physical examination revealed no cardiac murmur, no signs of pulmonary congestion, no peripheral edema, and no clinical evidence of overt heart failure.
Initial laboratory investigations were within normal limits. Complete blood count showed hemoglobin 14.2 g/dL, white blood cell count 7.8 × 109/L, and platelet count 245 × 109/L. Serum electrolyte analysis showed sodium 139 mmol/L, potassium 4.2 mmol/L, chloride 102 mmol/L, calcium 9.3 mg/dL, and magnesium 2.0 mg/dL. Renal function tests were normal, with serum creatinine 0.8 mg/dL and blood urea nitrogen 14 mg/dL. Random blood glucose was 96 mg/dL. Cardiac biomarkers were not elevated, with troponin I <0.01 ng/mL at presentation and no significant rise on repeat testing. Liver function tests and coagulation profile were also within normal limits.
A 12-lead electrocardiogram demonstrated widespread ST-segment depression in the inferolateral leads with reciprocal ST-segment elevation in lead aVR, raising concern for diffuse subendocardial ischemia rather than a localized coronary territory infarction (Figure 1). No sustained ventricular arrhythmia was documented on the initial electrocardiogram. Continuous cardiac monitoring showed sinus tachycardia without documented sustained ventricular tachycardia before cardiac arrest. Twelve-lead electrocardiogram showing diffuse ST-segment depression in the inferolateral leads with ST-segment elevation in lead aVR, consistent with global subendocardial ischemia.”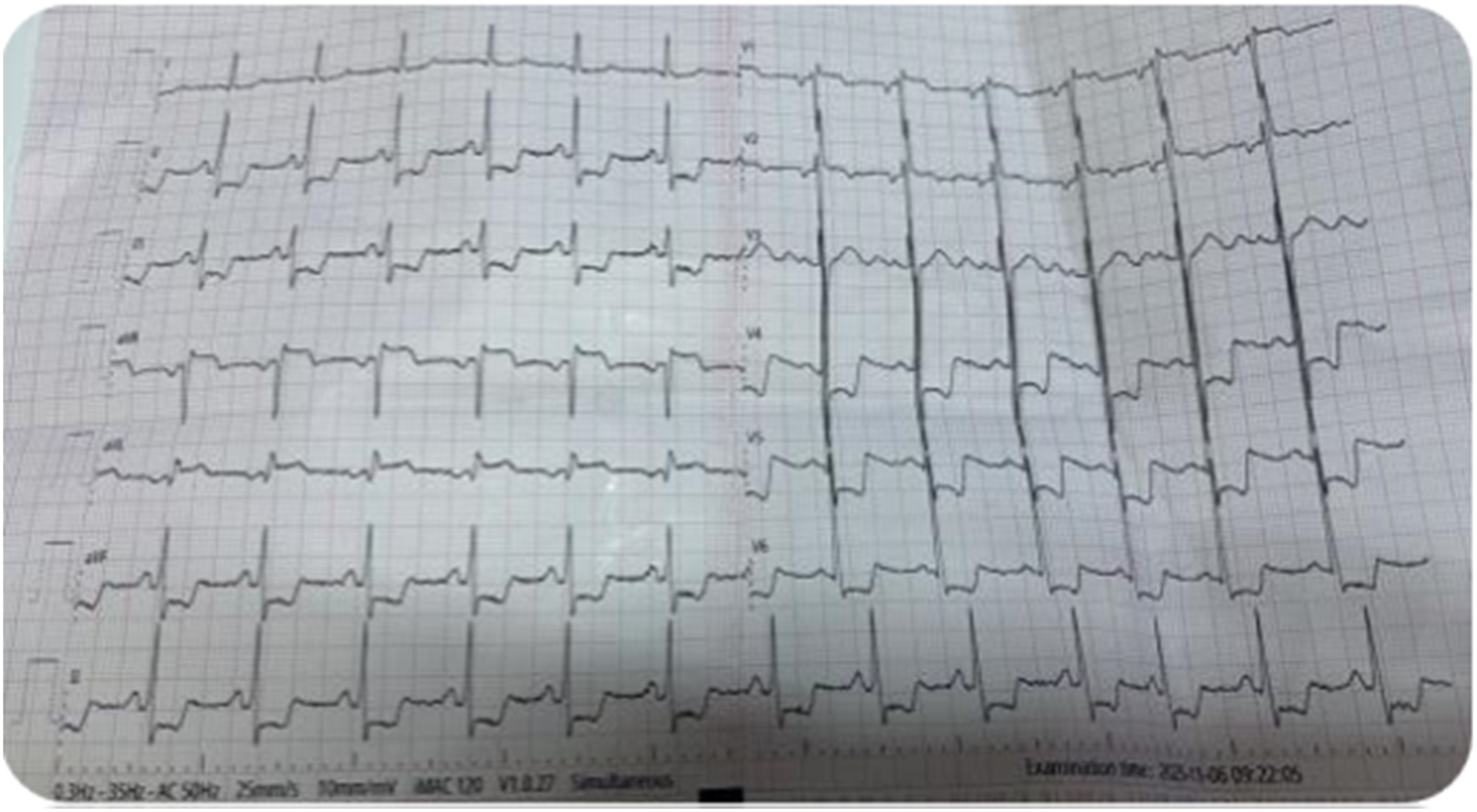
Transthoracic echocardiography revealed preserved left ventricular systolic function, with an estimated left ventricular ejection fraction of 65%. Right ventricular size and systolic function were preserved. The main pulmonary artery was mildly dilated, and mild pulmonary regurgitation was noted. No significant regional wall motion abnormality was identified. There was no pericardial effusion, intracardiac thrombus, or major valvular abnormality.
Given the recurrent syncope, hemodynamic instability, and high-risk ischemic electrocardiographic pattern, the patient was managed as a high-risk cardiac case. Initial management included oxygen supplementation as needed, intravenous fluid support, continuous cardiac monitoring, and urgent cardiology evaluation. Coronary computed tomography angiography was not available at our center. Therefore, urgent invasive coronary angiography was performed to evaluate the coronary anatomy and exclude obstructive coronary artery disease.
The clinical timeline was as follows: the patient presented to the emergency department following a prolonged syncopal episode. Initial assessment, including vital signs, physical examination, 12-lead electrocardiography, and laboratory investigations, was completed shortly after arrival. Transthoracic echocardiography was performed approximately 2 hours after presentation. In view of persistent hemodynamic instability and high-risk ischemic electrocardiographic findings, urgent invasive coronary angiography was performed approximately 4 hours after presentation.
Coronary angiography revealed an anomalous origin of the right coronary artery from the pulmonary artery. Retrograde flow was observed from the right coronary artery into the pulmonary trunk, consistent with a significant coronary steal phenomenon (Figure 2). No obstructive atherosclerotic coronary artery disease was identified. These findings established the diagnosis of anomalous origin of the right coronary artery from the pulmonary artery and provided a likely explanation for the patient’s recurrent syncope and diffuse ischemic electrocardiographic changes. Coronary angiography demonstrating anomalous origin of the right coronary artery from the pulmonary artery (ARCAPA). (A) Left coronary angiography showing a dilated left coronary system with extensive intercoronary collateralization. (B) Late phase imaging demonstrates retrograde opacification of the right coronary artery through collaterals, followed by drainage of contrast into the main pulmonary artery. (C) Absence of a right coronary ostium arising from the aorta with retrograde filling of the RCA, confirming ARCAPA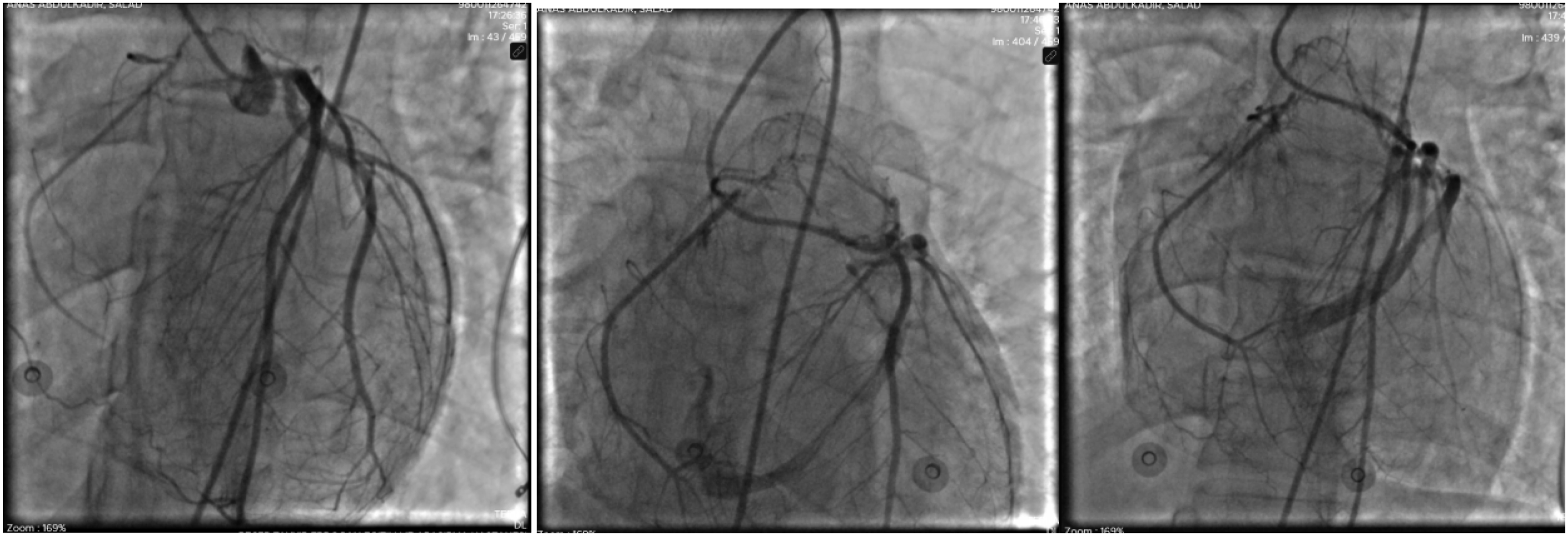
Approximately 30 minutes after coronary angiography, the patient developed sudden hemodynamic collapse and progressed to cardiac arrest. The initial arrest rhythm was not definitively documented, although malignant ventricular arrhythmia was clinically suspected. Advanced cardiopulmonary resuscitation was initiated immediately according to advanced life support protocols. Resuscitative measures included high-quality chest compressions, airway support, adrenaline administration, repeated rhythm assessment, and defibrillation if indicated. Despite prolonged resuscitative efforts lasting approximately 45 minutes, return of spontaneous circulation was not achieved, and the patient was pronounced dead.
The presumed mechanism of death was malignant ventricular arrhythmia triggered by severe myocardial ischemia related to ARCAPA and coronary steal phenomenon. However, because complete procedural monitoring data and terminal rhythm documentation were unavailable, the exact mechanism of death could not be definitively established. No post-mortem examination was performed.
Discussion
Anomalous origin of the right coronary artery from the pulmonary artery (ARCAPA) is an exceptionally rare congenital coronary anomaly, with most available evidence derived from isolated case reports, small case series, and systematic reviews. Although ARCAPA has often been regarded as less clinically severe than anomalous origin of the left coronary artery from the pulmonary artery, it is not a benign condition. Reported presentations range from incidental diagnosis to myocardial ischemia, heart failure, ventricular arrhythmias, and sudden cardiac death.1-3
The present case is clinically important because it demonstrates a fatal presentation of ARCAPA in a young adult with recurrent syncope, diffuse ischemic electrocardiographic changes, preserved ventricular systolic function, and angiographic evidence of coronary steal. This combination is notable because preserved ventricular function may falsely suggest lower risk. However, as shown in this case, normal systolic function does not exclude significant ischemic burden or vulnerability to malignant arrhythmias.
The pathophysiology of ARCAPA is mainly explained by abnormal coronary perfusion and coronary steal. After birth, pulmonary arterial pressure and pulmonary vascular resistance decrease. As a result, blood may flow retrogradely from the left coronary system through collateral vessels into the right coronary artery and then drain into the low-pressure pulmonary artery. This creates a left-to-right shunt at the coronary level and reduces effective myocardial perfusion. The resulting coronary steal may cause chronic or intermittent myocardial ischemia, particularly during exertion, tachycardia, hypotension, or other states of increased myocardial oxygen demand.2,3
This mechanism may also explain why the clinical presentation of ARCAPA is variable. Some patients remain asymptomatic for years because collateral circulation partially maintains myocardial perfusion. Others develop symptoms when collateral flow becomes insufficient or when the steal phenomenon becomes hemodynamically significant. Therefore, risk in ARCAPA depends not only on ventricular function but also on the degree of collateralization, direction and magnitude of coronary flow, myocardial oxygen demand, ischemic burden, and arrhythmogenic substrate.
Recurrent syncope should be considered a high-risk symptom in patients with ARCAPA. In young patients, syncope is frequently attributed to benign causes; however, syncope that occurs repeatedly, occurs during exertion, occurs without prodromal symptoms, or is associated with ischemic electrocardiographic abnormalities should raise concern for a cardiac etiology. In this case, recurrent syncope may have represented transient global myocardial ischemia or self-terminating ventricular arrhythmias before the final fatal event. Therefore, syncope in ARCAPA should not be interpreted as benign, especially when accompanied by evidence of myocardial ischemia.
The electrocardiographic findings in this patient were also important. Diffuse ST-segment depression with ST-segment elevation in lead aVR is commonly associated with global subendocardial ischemia and is classically described in severe left main coronary artery disease or multivessel coronary artery disease.9,10 However, this pattern is not specific to atherosclerotic coronary disease and may occur in other conditions that produce diffuse myocardial oxygen supply-demand mismatch. In the present case, the absence of obstructive coronary artery disease and the angiographic demonstration of retrograde flow from the right coronary artery into the pulmonary artery support ARCAPA-related coronary steal as the likely mechanism of global ischemia.
Multimodality imaging is central to the diagnosis and management of congenital coronary artery anomalies. Transthoracic echocardiography may provide initial clues, including abnormal coronary flow, dilated coronary arteries, pulmonary artery abnormalities, or associated structural findings. Coronary computed tomography angiography is particularly useful because it provides detailed anatomical definition of the anomalous coronary origin, course, and relationship to surrounding structures. Cardiac magnetic resonance imaging may further assess ventricular function, myocardial viability, perfusion, and scar. Invasive coronary angiography remains valuable for demonstrating collateral circulation and retrograde drainage into the pulmonary artery. 8
In the present case, coronary computed tomography angiography was unavailable, and invasive coronary angiography was required to establish the diagnosis. Angiography demonstrated anomalous origin of the right coronary artery from the pulmonary artery with retrograde flow into the pulmonary trunk, confirming a significant coronary steal phenomenon. The patient’s sudden deterioration shortly after angiography highlights the fragile hemodynamic state that may exist in symptomatic ARCAPA. Although the exact mechanism of arrest could not be definitively established, possible contributors include severe myocardial ischemia, malignant ventricular arrhythmia, hemodynamic instability, or procedure-related stress in an already ischemic myocardium.
Management of ARCAPA should not be limited to diagnosis. Surgical correction is generally recommended after diagnosis, including in asymptomatic patients, because of the persistent risk of myocardial ischemia, ventricular arrhythmia, and sudden cardiac death.3,7,8 The preferred surgical approach is usually direct reimplantation of the anomalous right coronary artery into the aorta, which restores a two-coronary-artery circulation and eliminates the coronary steal. Alternative strategies include ligation of the anomalous origin combined with coronary artery bypass grafting, particularly when direct reimplantation is not technically feasible. The choice of surgical technique depends on coronary anatomy, patient condition, myocardial viability, and surgical expertise.
The fatal outcome in this case underscores the need for early recognition and urgent surgical referral. Earlier diagnosis may have allowed transfer to a center with congenital cardiac surgical capability and definitive repair before clinical deterioration. Although it cannot be proven that earlier intervention would have prevented death, this case supports treating symptomatic ARCAPA, especially when associated with syncope and ischemic electrocardiographic changes, as a high-risk condition requiring expedited management.
This case has important implications for clinicians, particularly in resource-limited settings. Young patients with recurrent unexplained syncope and ischemic electrocardiographic abnormalities should be evaluated for structural and congenital coronary abnormalities, even when ventricular systolic function is preserved and traditional cardiovascular risk factors are absent. ARCAPA should be included in the differential diagnosis because delayed recognition may result in sudden and irreversible deterioration.
Limitations
This case report has several limitations. First, complete procedural and monitoring data at the time of cardiac arrest were not available. Although malignant ventricular arrhythmia was clinically suspected as the most likely cause of death, the initial arrest rhythm and terminal rhythm were not definitively documented. Therefore, the exact mechanism of cardiac arrest could not be confirmed.
Second, coronary computed tomography angiography was not available at our center. This limited early noninvasive anatomical characterization of the anomalous coronary artery, including its precise origin, course, and relationship to adjacent structures. Although invasive coronary angiography confirmed the diagnosis of ARCAPA and demonstrated retrograde flow into the pulmonary artery, coronary computed tomography angiography could have provided additional anatomical detail for surgical planning.
Third, no post-mortem examination was performed. As a result, pathological confirmation of the cause of death was not possible. Other potential contributing factors, including severe myocardial ischemia, procedure-related hemodynamic instability, contrast-related effects, or an undocumented arrhythmia, could not be completely excluded.
Fourth, this is a single case report; therefore, the findings cannot be generalized to all patients with ARCAPA. The fatal course observed in this patient may not reflect the clinical outcome of patients diagnosed earlier or treated surgically before the development of severe ischemic or arrhythmic complications.
Finally, some clinical details were limited by the emergency nature of the presentation and the patient’s rapid deterioration after angiography. Despite these limitations, this case highlights the potential for sudden fatal deterioration in symptomatic ARCAPA, even when left ventricular systolic function is preserved.
Conclusion
ARCAPA is a rare but potentially fatal congenital coronary artery anomaly. This case highlights that recurrent syncope in a young patient should not be dismissed, particularly when associated with ischemic electrocardiographic abnormalities. Diffuse ST-segment depression with ST-segment elevation in lead aVR may reflect global subendocardial ischemia and should prompt urgent evaluation for both atherosclerotic and non-atherosclerotic causes, including congenital coronary anomalies.
Preserved ventricular systolic function does not exclude significant ischemic or arrhythmic risk in ARCAPA. Early recognition, multimodality imaging, close monitoring, and urgent referral for surgical correction are essential to prevent sudden cardiac death. This case emphasizes the narrow window for diagnosis and intervention in symptomatic ARCAPA.
Footnotes
Ethical Considerations
The study protocol was reviewed and approved by the Institutional Review Board (IRB) of Mogadishu Somali-Turkish Training and Research Hospital. The study was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki.
Consent for Publication
Written informed consent for publication of this case report and accompanying anonymized clinical data and images was obtained from the patient’s legal next of kin after the patient’s death.
Author Contributions
Funding
The authors received no financial support for the research, authorship, or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors declare that there are no conflicts of interest regarding the publication of this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article. Additional information may be obtained from the corresponding author upon reasonable request.