
Abstract
The XLH Matters 2024 GCC Edition meeting convened 51 physicians from 6 Gulf countries to discuss the diagnosis and management of patients with X-linked hypophosphatemia (XLH). This was the first XLH Matters meeting held in the Gulf Cooperation Council (GCC) region, reflecting the unique healthcare structure, cultural context, and patient needs. The key themes of the meeting included: challenges faced by GCC clinicians in diagnosing and treating XLH, the importance of multi-disciplinary care, the psychosocial impact, and the principles of effective transition of patients from pediatric to adult care. Participants emphasized the importance of raising awareness of XLH among primary care physicians, pediatricians, and dentists to facilitate early diagnosis in the region. Genetic testing was highlighted as the key tool supporting diagnosis, but wider access to this is needed across the GCC. A structural, progressive, and personalized model for pediatric-to-adult transition was proposed, based on the individual needs of the patient and including patient empowerment and continuity of care. The meeting underscored the need for regional collaboration, awareness initiatives, and implementation of recent clinical guidelines to improve outcomes for people with XLH in the GCC region.
Plain Language Summary
The XLH Matters 2024 GCC Edition meeting brought together 51 doctors from 6 Gulf Cooperation Council (GCC) countries to discuss how to better diagnose and manage people living with X-linked hypophosphataemia (XLH). This was the first time the XLH Matters meeting had been held in the Gulf region, providing an opportunity to address the area’s specific healthcare systems, cultural settings, and patient needs. During the meeting, doctors explored several important topics: the challenges of identifying and treating XLH in the GCC, the value of multidisciplinary care, the emotional and social impact of the condition, and the importance of supporting patients as they move from child to adult healthcare services. Participants agreed that more awareness is needed among primary care doctors, pediatricians, and dentists so that XLH can be recognized and treated earlier. They also highlighted the role of genetic testing in confirming a diagnosis of XLH, while acknowledging that access to testing should be improved across the region. A structured and personalized plan for moving patients from pediatric to adult care was proposed. This approach would focus on the individual needs of each person, continuity of care, and empowering patients to manage their condition confidently. The meeting concluded with a call for stronger collaboration within the region, more education and awareness programs, and better adoption of international clinical guidelines. Together, these steps can help improve care and quality of life for people living with XLH in the Gulf region.
Keywords
Introduction to XLH Matters
The XLH Matters meetings have become an important and recurring mainstay forum for healthcare professionals (HCPs) who treat patients with X-linked hypophosphatemia (XLH). They provide an opportunity for networking, dialog, and the sharing of real-world data. XLH Matters also establishes a platform for clinicians to form cross-disciplinary networks and share recommendations arising from their clinical experience.
Past XLH Matters meetings were held in Frankfurt (2023) and Madrid (2022 & 2024), with their summaries published previously.1,2 These meetings have demonstrated a real-world impact on patient care and quality of life (QoL). For example, a Gulf Cooperation Council (GCC)-based adult endocrinologist reported the successful diagnosis and treatment with burosumab of a 15-year-old patient referred by an orthopedist, based on their familiarity with features of the condition gained from attending a previous XLH Matters meeting. This paper complements previous XLH Matters reports by providing the first detailed account of GCC-specific challenges, local experience-based practical solutions, and a proposed regional transition model adapted to local healthcare systems.
XLH Matters 2024 GCC Edition
The present meeting was the first XLH Matters held in the GCC, a regional organization comprising 6 Middle Eastern countries (Bahrain, Kuwait, Oman, Qatar, Saudi Arabia [KSA], and the United Arab Emirates [UAE]). Based in the Arabian Peninsula, the GCC countries share common goals and cultural ties, with a total population of 61.5 million in 2024. The GCC is a region with a distinct culture, population, and healthcare infrastructure. Large extended families with multiple affected members, high consanguinity rates, overlapping nutritional rickets, and uneven access to specialist services, advanced diagnostics, and therapeutics all create specific challenges for XLH recognition, treatment initiation, and lifelong follow-up.3,4 At the same time, emerging XLH registries and growing regional expertise offer opportunities to standardize care and implement the latest international best practice.3,4 These distinctions present both challenges and opportunities in the treatment of patients with XLH in the region.
XLH Matters 2024 GCC Edition was designed to address these challenges, with key objectives listed in Box 1. For the first time outside of Europe, XLH Matters brought together 51 physicians from 6 GCC countries for a 2-day in-person meeting to share regional knowledge and best practice, through a series of presentations, workshops, and discussions (Table 1). Specialists treating both pediatric and adult patients attended the meeting.
Key Objectives of XLH Matters 2024 GCC Edition.

XLH Matters 2024 GCC Edition – Workshops & Discussions.

Abbreviations: GCC, Gulf cooperation council; XLH, X-linked hypophosphatemia.
Methodology
For each workshop, participants were assigned to moderated roundtable groups (6-10 clinicians per table). Moderators used pre-defined prompts and structured question guides to focus discussion on diagnostic challenges, treatment decision-making, MDT composition, and transition of care. Key discussion points, areas of agreement, and examples of regional practice were captured in real time by medical writers using standardized note-taking templates and were cross-checked against audio recordings where available. After the meeting, the faculty reviewed the collated outputs in an iterative process, identified areas of convergence, and agreed by informal consensus on the themes and practical recommendations reported here. No formal quantitative voting or Delphi process was conducted.
Background of XLH
In normal bone and tooth development, calcium and phosphate combine to form hydroxyapatite crystals, which are deposited by osteoblasts and ameloblasts into the collagen matrix to provide structural strength and mineralization. 5 Fibroblast growth factor 23 (FGF23) helps maintain balanced mineralization by reducing renal phosphate reabsorption and lowering active vitamin D levels, ensuring calcium-phosphate deposition into hydroxyapatite occurs in a tightly regulated, physiologic range during normal development.5 -7 XLH is a rare inherited genetic disorder characterized by renal phosphate wasting that arises from variants in the PHEX (phosphate-regulating endopeptidase homolog, X-linked) gene on the X chromosome. 5 Inactivation of the PHEX gene leads to elevated production of FGF23, which, when released into the circulation, binds to the Klotho-fibroblast growth factor receptor 1 (FGFR1) in the proximal tubule of the kidneys, inducing hyperphosphaturia and chronic hypophosphatemia (See Figure 1).6,7 The resultant phosphate wasting leads to a range of clinical manifestations, most commonly presenting in childhood as lower leg deformities. 8 XLH is a lifelong, multisystemic disease, with manifestations presenting and accumulating through different stages of life. In childhood, XLH can cause progressive symptoms including rickets, osteomalacia, muscle weakness, bone pain, and dental abscesses.7,8 Adults with XLH may also be affected by fractures, pseudofractures, osteoarthritis with osteophytes, hearing loss, dental abscesses, spinal stenosis, enthesopathy, and numerous musculoskeletal symptoms that affect mobility. 7

Pathophysiology of XLH and mechanism of action of burosumab.
Diagnostic Challenges
The rarity of XLH combined with its variable presentation creates significant diagnostic challenges. Delayed diagnosis and misdiagnosis of XLH is a major obstacle in disease management and is linked to poor patient outcomes due to the progression of symptoms. 9 The impact of misdiagnosis in childhood impacts adult and geriatric bone health, and is associated with increased fracture risk, complex comorbidity, impaired quality of life, substantial burden on the patient, family, and caregivers, and increased healthcare costs. 9 Clinicians (including dentists and nurses) outside specialist centers may lack knowledge of XLH, causing missed or incorrect diagnoses and delays in referring patients to the appropriate MDT team. Moreover, misconceptions that XLH affects only children lead to awareness being concentrated primarily among pediatricians. As such, adult patients may struggle to gain access to appropriate care outside of specialist centers. 10 Differentiating XLH from nutritional or other genetic forms of rickets is challenging, especially in the GCC region, where vitamin D deficiency and high consanguinity rates increase the prevalence of alternative causes of rickets.11 -14
Multidisciplinary Care
Increased physician awareness is needed to reduce diagnostic delay, and a strong multidisciplinary clinician network is vital for optimal lifelong XLH patient care.2,15 After diagnosis, patients with XLH require multidisciplinary care from a range of physicians and allied specialists to manage the multisystemic nature of the disease. 15 This is ideally organized by an expert in metabolic bone disease and provided by a team of specialists including endocrinologists, radiologists, orthopedic surgeons, nephrologists, physical therapists, rheumatologists, dentists, and other specialties, depending on individual patient needs. 15 Care for patients often extends to specialists such as neurosurgeons, ENT surgeons, and orthodontists. The psychological impact of XLH evolves over the disease course and includes increased risk of depression and body image concerns, which often requires psychological support from mental health professionals, e.g., social workers, psychiatrists, and psychologists.16,17
Treatment of XLH
Pharmacological treatment for XLH comprises either supplementation with oral phosphate and active vitamin D 18 or burosumab, a human IgG1 monoclonal antibody which inhibits the activity of FGF23. 15 With oral phosphate and active vitamin D, the short biological half-life of inorganic phosphate necessitates multiple daily doses for up to 6 times per day. 19 Moreover, long-term treatment with oral phosphate and active vitamin D is associated with endocrine complications, including secondary and tertiary hyperparathyroidism, as well as renal adverse events such as hypercalciuria and nephrocalcinosis. 17 Frequent dosing, poor taste, and side effects often lead to poor adherence, resulting in incomplete healing of rickets and osteomalacia and the need for corrective surgery for limb deformities. In contrast, burosumab has been shown to be superior in promoting healing of rickets and osteomalacia. 20 Inadequate treatment can also contribute to pseudofractures and chronic pain.7,19 Burosumab occupies a key place in the treatment of XLH in the Gulf region; it has been approved by regulatory authorities for the treatment of XLH and tumor-induced osteomalacia (TIO) in all GCC countries. 21
Updated clinical guidelines on the diagnosis and treatment of patients with XLH were published recently by Haffner and colleagues 15 and the International Working Group.22,23 With continual advancements in XLH management, networking meetings, such as XLH Matters, provide a key platform for the sharing of regional knowledge and the discussion of real-world data that inform clinical practice.
XLH Matters 2024 GCC Edition – Meeting Sessions
The XLH Matters 2024 GCC Edition meeting comprised three roundtable workshops and a series of interactive discussions led by expert faculty. A summary of the meeting is provided in Table 1.
Pediatric Workshop – Identifying XLH
The pediatric workshop was preceded by an introductory presentation on common difficulties in diagnosing XLH in children. The participants then formed roundtable discussion groups and addressed the challenges encountered in their clinics and how these could be overcome.
Introduction
The variable pathophysiology of rickets is a major challenge in the differential diagnosis of XLH in the GCC region. In patients presenting with symptoms of rickets and low phosphate, XLH may appear a likely diagnosis. Eventually, almost all cases of rickets present with hypophosphatemia even though this may not be the primary cause of compromised mineralization (see Figure 2), and it can be challenging to differentiate XLH-related hypophosphatemia from other etiologies. 24

Rickets has numerous etiologies, all of which result in chronic hypophosphatemia.
Differential Diagnosis of XLH
Rickets can be either calcipenic (associated with elevated parathyroid hormone [PTH] levels) or phosphopenic (see Figure 1). Both types are associated with renal phosphate wastage. 25 Elevated PTH in calcipenic rickets may be due to dietary calcium/vitamin D deficiency, impaired renal processing of 25(OH)D, and end-organ resistance to the active form of vitamin D.24,25 Non-XLH causes of phosphopenic rickets include primary and secondary Fanconi syndrome or variants in genes other than PHEX (e.g. FGF23, DMP1, ENPP1, FAM20C, or SLC34A3; Table 2). 24
Common Genetic Variants a Causing Phosphopenic Rickets.

Abbreviations: AD, autosomal dominant; AR, autosomal recessive; FGF23, fibroblast growth factor 23; XL, X-linked.
This is a non-exhaustive list. For a more detailed list, see https://www.genomicseducation.hee.nhs.uk/genotes/knowledge-hub/hereditary-hypophosphataemic-rickets/
Pediatric Clinical Complications of XLH
XLH frequently presents with multiple clinical manifestations, and challenges may arise during clinical investigations of pediatric patients with suspected rickets. Pediatric patients presenting with bowed legs (genu varum) or knock knees (genu valgum) require detailed investigation to determine if these are a result of normal variation in growth, an alternate etiological cause such as Blount’s disease or a skeletal dysplasia, such as Schmid metaphyseal chondrodysplasia, or XLH. Dental symptoms are common among pediatric XLH patients which may affect deciduous and permanent teeth due to impaired mineralization of dentine and enamel, allowing bacteria to invade the pulp, leading to dental abscess in the absence of trauma or tooth decay. 8 Dental manifestations should always be investigated, 15 and suspected XLH must be differentiated from dental enamel hypoplasia resulting from nutritional rickets. A family history of rickets should also be scrutinized to determine whether another autosomal disorder 15 or an X-linked recessive disease, such as Dent’s or Lowe’s disease, may be the cause of rickets.
Biochemical Profile of XLH
Biochemically, XLH is characterized by renal phosphate wasting which results in hypophosphatemia. In pediatric patients, interpretation of serum phosphate must account for age-related reference ranges as normal values are higher in children than in adults. 26 Calculating the tubular maximum reabsorption of phosphate per glomerular filtration rate (TmP/GFR) is essential for estimating renal phosphate loss. 27 TmP/GFR is an index of the renal tubular threshold for phosphate reabsorption and is derived from paired serum and urine phosphate and creatinine measurements, with suitable adjustments for age and sex. 27 To rule out calcipenic rickets, clinicians should assess PTH activity and vitamin D levels. 27 Measuring FGF23, preferably using intact FGF23 assays, is important as increased FGF23 is a hallmark of XLH. 15
Imaging Profile of XLH
Radiologically, XLH in growing pediatric patients is characterized by metaphyseal widening, cupping, and fraying. 25 The trabecular pattern at the distal ends of long bones often shows a sclerotic appearance. 21 In older adolescents and adults, common findings include pseudofractures, osteoarthritis, osteophyte formation, calcification of ligaments and joint capsules, and spinal stenosis. 15 Renal ultrasound may reveal nephrocalcinosis in patients receiving active vitamin D plus phosphate. 8
Genomics
Genetic analysis is critical for identifying XLH in pediatric patients. Targeted next-generation sequencing (NGS) gene panels allow accurate identification of underlying mutations and aid in differential diagnosis. 28 Whole-exome sequencing (WES) or whole-genome sequencing (WGS) are powerful NGS that may be employed to diagnose complex or atypical XLH cases, but they vary in scope, diagnostic utility, and cost.21,29 In brief, WGS sequences the entire genome, including both protein-coding and non-coding regions, while WES focuses only on protein-coding regions, offering a more affordable and accessible option but potentially missing rare, high-impact variants found outside the exome.21,29 The diagnosis of XLH as the cause of HR ultimately involves careful consideration of a range of clinical, biochemical, radiological, and genetic factors, in combination with the evaluation of family history as a key diagnostic indicator. 15
Workshop Discussion – Challenges in Identifying XLH in Pediatric Patients
Pediatric care providers were grouped and asked to consider the challenges they have encountered when diagnosing pediatric patients and the potential solutions to improve early diagnosis.
Challenges in Identifying XLH in Pediatric Patients
Participants agreed that delayed diagnosis of XLH was the major challenge when receiving pediatric patients. This is driven by a lack of awareness of XLH among referring physicians, who may have a low index of suspicion regarding rickets-like symptoms and the atypical manifestations of the disease. Atypical cases of XLH further drive diagnostic delays and exacerbate the issue of awareness, since they require even more comprehensive education to inform physicians of the diversity of possible manifestations. Classic signs of XLH such as dental abscesses can be missed by physicians who are unfamiliar with XLH. Increased education around XLH is needed, 1 not only among pediatric endocrinologists but also general pediatricians and pediatric orthopedic surgeons who are the likely source of referral to specialists. Improving disease awareness among physicians and patients would likely lead to earlier diagnosis and initiation of appropriate treatment. 30
Attendee quotation
• “We need to raise the index of suspicion among family physicians, dentists, and pediatricians.”
• “The general public and families need to be aware of XLH because if they are aware, they will seek advice.”
Considering the frequent delayed diagnosis of XLH, the attendees highlighted several “red flags” to raise diagnostic suspicion, presented in Box 2.
Summary of Key Indicators for Differential Diagnosis of XLH in Pediatric Patients.
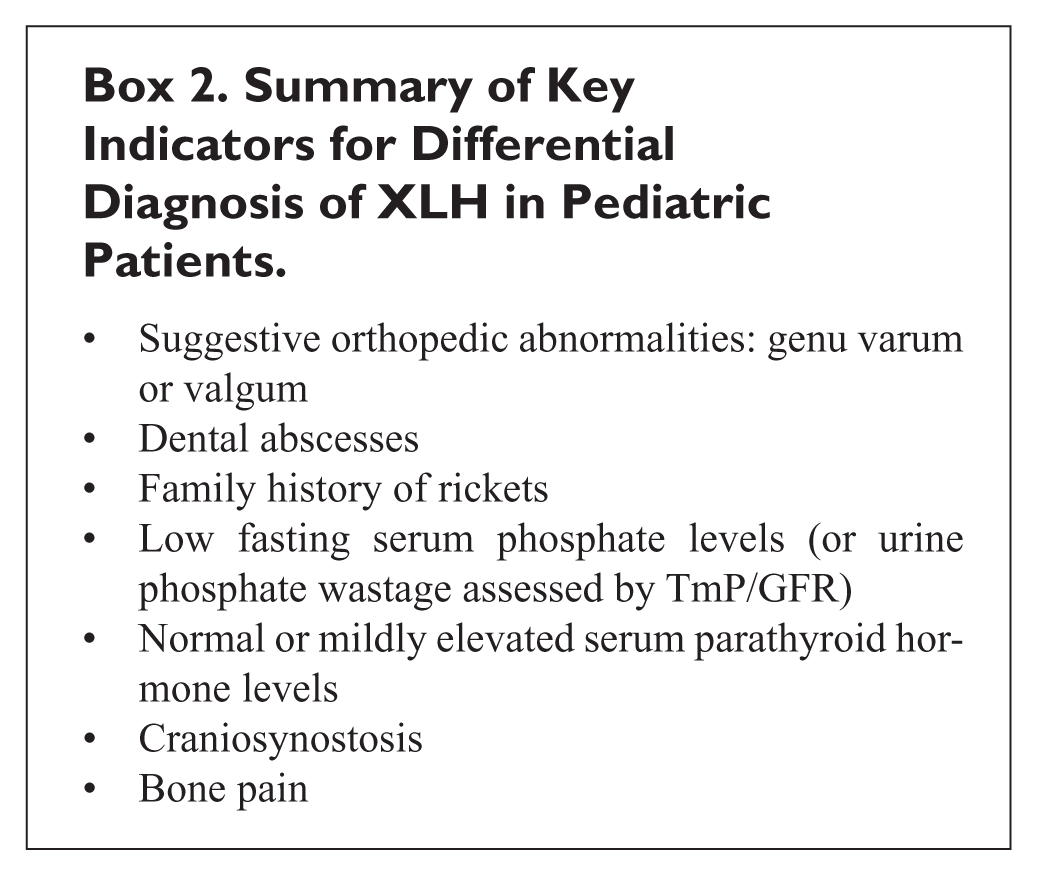
Red Flags for XLH Diagnosis
Diagnosis of XLH can be delayed when not supported by a complete family history. Collecting family history supports clinicians in distinguishing between cases of heritable rickets and those resulting from de novo PHEX mutations. Family history also provides insight into inheritance patterns for differential diagnosis of rickets (e.g. autosomal dominant vs autosomal recessive vs X-linked recessive). 15 Evidence has shown that if 1 member of a family is diagnosed with XLH, there may be at least 1 other affected family member. 3 There is also a risk of losing patients to clinical follow up due to a perception of poor efficacy with oral phosphate and active vitamin D or limited access to effective treatment. The lack of knowledge of XLH is, therefore, a challenge which extends beyond physicians to XLH patients themselves and their extended families. Insufficient education surrounding XLH among patients and caregivers contributes to delayed diagnosis and consequently poorer outcomes for XLH patients. 9
Genetic Testing and Regional Challenges
Genetic testing was highlighted as a key tool to support early identification of XLH in pediatric patients, and diagnosing XLH without the results of genetic testing was reported to be very challenging and comes with the risk of starting inadequate treatment. However, genetic testing may be hampered by lack of availability and/or cost. Further, it was pointed out that FGF23 testing is not readily available in all GCC centers, and the waiting time for genetic testing results can be significant. The reliability of FGF23 testing in the GCC region was reported to be not always consistent. This could be addressed by collaboration with larger centers that have access to genetic testing and referring patients to the Gulf FGF23-related HR and Osteomalacia Registry. 31
Vitamin D Testing and Regional Challenges
Vitamin D is derived predominantly from cutaneous synthesis, where 7-dehydrocholesterol is photoconverted by solar UVB radiation to cholecalciferol (vitamin D3), with smaller contributions from dietary sources. Cholecalciferol is subsequently hydroxylated in the liver to 25-hydroxyvitamin D and then undergoes 1α-hydroxylation in the kidney to form 1,25-dihydroxyvitamin D (calcitriol), the biologically active hormone regulating calcium and phosphate homeostasis.
Limited access to 1,25-dihydroxyvitamin D testing in the GCC complicates the diagnosis of XLH. Attendees noted that many GCC centers lack validated age- and gender-adjusted biochemical reference ranges (particularly fasting phosphate) making interpretation challenging. Obtaining fasting blood and urine samples from pediatric patients can be logistically difficult, which may hinder biochemical assessment. Calculated TmP/GFR requires fasting samples, which can be difficult to collect from young children. Standardized and reference ranges for FGF23 assays for pediatric patients remain incompletely established. As an alternative, for phosphate TmP/GFR assessment, spot urine and dip-stick methods have been recommended as they are simpler to perform for young patients. 15
Summary
In the GCC, identifying pediatric XLH patients is hindered by low awareness among clinicians and limited access to diagnostic tools. Biochemical and genetic testing may be costly or unavailable, and accurate age- and gender-specific reference ranges for biochemical variables are necessary for interpretation. Pediatric testing challenges, such as with TmP/GFR, highlight the need for child-friendly assays. Guidelines now recommend non-fasting serum phosphate assessments with repeated measurements in equivocal cases. 13
Adult Workshop – When and How to Treat XLH in the GCC
Adult-treating physicians took part in the adult workshop, which ran in parallel with the pediatric workshop. Grouped attendees considered case studies that illustrated specific challenges in the treatment of adult patients. focusing on practical decision points regarding when to initiate treatment and how to choose between burosumab and conventional oral phosphate plus active vitamin D. Participants discussed clinical triggers for treatment (e.g. pseudofractures, progressive pain, impaired mobility, or biochemical osteomalacia), factors influencing the choice of therapy, and strategies for ongoing monitoring and dose adjustment.
Workshop Discussion – Practical Guidance on the Common Challenges in the Management of Adult XLH Patients in the GCC Region
Managing adults with XLH comes with challenges related to existing skeletal deformation, comorbidities, and the potential for masking symptoms through lifestyle modifications.
Red Flags for an XLH Diagnosis
Adult patients may go undiagnosed if their symptoms are mild, particularly if they have adapted to avoid situations that cause pain. Adults with XLH often adapt by subtly modifying their physical activity, such as switching to low-impact movements, reducing physical activity, or pacing tasks to manage chronic fatigue. Many also adopt environmental or practical adaptations, such as mobility aids avoid situations that exacerbate symptoms.7,40 When assessing such patients, clinicians should question them about their lifestyle to uncover any such adaptations. Patients who complain of pain in the thigh/upper leg should raise a flag for potential pseudofractures, which are easily identified on X-ray by an experienced radiologist. However, clinicians are advised to also check X-rays themselves to ensure they are not missed, especially since burosumab has shown good efficacy for pseudofracture healing in adults.32 -34 Further, the presented case study evidence highlighted the successful use of burosumab for the treatment of a 67-year-old female with progressive immobilizing joint pain and severe osteoarthritis, who became ambulant without crutches 6 months after treatment initiation.
Attendee quotation
• “Before starting treatment in adults, we must define treatment goals to evaluate success.”
Secondary Hyperparathyroidism
Secondary hyperparathyroidism (HPT) is a frequent complication encountered in up to a quarter of adult XLH patients, 35 particularly among those who are receiving long-term phosphate supplementation. 15 HPT is driven by low 25-hydroxyvitamin D (25[OH]D) and high phosphate levels. In cases where calcium levels are normal or elevated, a positron emission tomography/computed tomography is advisable to look for parathyroid hyperplasia since the chronic suppression of 25(OH)D by FGF23 can trigger progressive polyclonal hyperplasia. If an adenoma is present and resectable, it is likely that this will be followed by a transient decrease in calcium and PTH levels. Patients with parathyroid adenoma may remain eligible for treatment with burosumab. Although burosumab normalizes phosphate levels in most cases, some patients experience persistent renal phosphate wasting even with PTH within the normal range; this is due to HPT that cannot be overcome through FGF23 inhibition alone. In such cases, functional assessments such as improvements in stiffness, pain and physical activity should be used to inform decisions on continued treatment with burosumab.
Iron Deficiency in XLH
Participants discussed iron deficiency, another common challenge in adults with XLH. Low iron levels increase FGF23 production which further exacerbates phosphate wasting. Exogenous iron in tablet/oral form allows the increased FGF23 to be cleaved and degraded. However, intravenous iron prevents FGF23 cleavage and can result in greater levels of intact FGF23 and thus phosphate wasting. In GCC practice, workshop participants concluded that iron status should be assessed in adults with XLH who have unexplained worsening of hypophosphatemia, and that iron deficiency should be corrected where present, preferably with oral preparations where feasible. Intravenous iron should be reserved for clear indications, with close biochemical monitoring, given its potential to aggravate phosphate wasting through sustained elevations in intact FGF23. 36
Attendee quotation
• “Some adults have strikingly abnormal biochemical profiles.”
If diagnosed in adulthood, XLH patients may have unusual biochemical profiles, reflecting the underlying multisystemic nature of the disease. Even if the patient is not taking burosumab or oral phosphate and active vitamin D, they should be monitored regularly for any changes in their condition that might reflect disease progression. If burosumab is initiated, dose changes up to a maximum of 90 mg can be considered as the patient progresses, preferably using an iterative approach to monitor the patient’s condition between doses. 37
GCC Regional Challenges
Several challenges specific to the GCC setting were highlighted. Access to intact FGF23 assays and timely genetic testing remains inconsistent across centers, which can delay confirmation of XLH diagnosis in adults who may present after years of untreated disease. Differences in reimbursement pathways for burosumab between countries and healthcare systems can influence whether adults receive burosumab or remain on, or revert to, oral phosphate and active vitamin D. In addition, the high prevalence of vitamin D deficiency and nutritional rickets in the region complicates interpretation of biochemical profiles in adults, reinforcing the need for careful evaluation of family history (including a pedigree analysis), prior childhood symptoms, and radiographic findings.
Summary
Diagnosing and treating adults with XLH is complex in part due to behavioral adaptation masking symptoms and resulting in a lack of clinical complaints. Clinicians in turn may overlook the underlying phosphate wasting and delay a formal diagnosis. Recent guidelines suggest adults with clear skeletal issues—like pseudofractures, radiological osteomalacia, severe pain, or functional impairments—should receive active treatment. Burosumab is recommended for pseudofractures or poor response to oral phosphate and active vitamin D. Where burosumab is not available or for milder cases in whom symptoms can be well controlled with active vitamin D and phosphate, such supplementation treatment is an option. 20 In the absence of clinical symptoms, clinicians may consider not starting treatment. Ongoing monitoring of symptoms, physical function, serum phosphate, and complications guides treatment decisions, dose adjustments, and therapy switches. HPT is a commonly encountered complication in the treatment of adult patients. Treatment decisions should be made following proper assessments to determine the origin of HPT. Adult patients with adenomas are candidates for burosumab treatment, but not all these patients will achieve normalized phosphate levels since phosphate wasting is mediated in some cases by HPT and cannot be overcome by burosumab dose increases. Functional assessments may be used in these adult patients to assess treatment efficacy.
In the GCC, challenges such as inconsistent access to FGF23 assays and genetic testing, varied burosumab reimbursement, and high rates of vitamin D deficiency and nutritional rickets complicate adult XLH diagnosis and management, necessitating thorough evaluation of family history, childhood symptoms, and radiographic findings.
Detailed diagnostic and treatment recommendations, including formal indications for burosumab and conventional therapy, are summarized in the “Updates to the international XLH guidelines” section.
Multidisciplinary Team Collaboration in the GCC
The necessity of MDT collaboration was outlined in an introductory presentation ahead of the MDT workshop. Owing to the multisystemic nature and variable manifestations of the disease, XLH patients require lifelong care from a multidisciplinary range of specialists. 15 This MDT should ideally be coordinated by a lead clinician with expertise in metabolic bone diseases, working with specialists as required. 15 The range of specialists may evolve over time depending on patient needs.
Coordinating an MDT for the treatment of XLH patients is accompanied by certain challenges. The rarity of the disease and lack of awareness among physicians 2 can make it difficult to locate specialists with experience of treating XLH. From the patients’ perspective, XLH is a challenging genetic disease characterized by considerable burden that requires lifelong management. 38 At present, many patients with XLH do not have access to appropriate healthcare and are, therefore, not properly diagnosed and treated. 4
Establishing effective MDT collaboration can result in increased patient satisfaction, more efficient use of resources, and improved health outcomes. 39 Current XLH clinical guidelines provide a schedule for the follow-up of patients across the range of specialists involved in XLH care, 15 which may serve as a basis for MDT collaboration.
Strengthening XLH Care Workshop – The Importance of a Multidisciplinary Approach
The importance of a multidisciplinary approach in the treatment of XLH and the key members of an MDT were discussed among pediatric and adult-treating attendees during the third workshop. Participants discussed the type of specialists essential for the diagnosis, treatment, and monitoring of XLH, as well as the challenges of establishing an MDT.
Workshop Discussion – Specialists Needed in the MDT
The key MDT specialists in pediatric and adult XLH management are summarized in Box 3.
Key Specialists Involved in the Diagnosis, Treatment, and Monitoring of XLH.

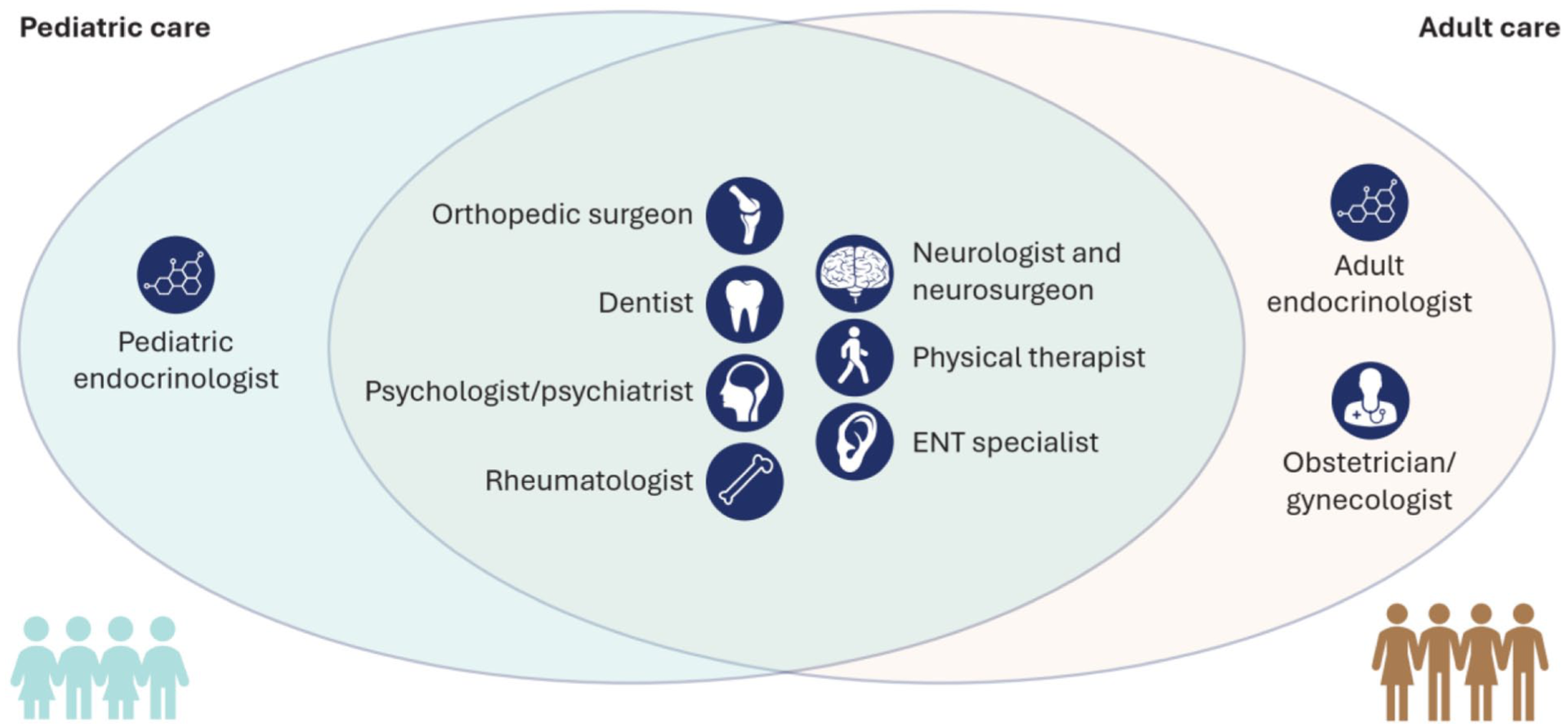
Participants also highlighted the importance of genetic counselors within the MDT who can support patients with their diagnosis and also counsel them on their heritability risks and options for family planning; the latter may be further supported by family planning specialists for adult patients. Moreover, a clinical pharmacist can explain the importance of medication to patients and facilitate the delivery of medication to remote locations.
Attendee quotation
• “Genetic counselors are important to support patients in having healthy children.”
Additional specialists may be necessary on a moderated basis to ensure individualized patient care. Dietitians can play a key role in promoting healthy eating and preventing obesity in XLH patients, who often have limited mobility from pain, joint stiffness, and osteoarthritis. Patients affected by significant pain symptoms should be assigned a pain management team to help alleviate rheumatological and bone pain.
The participants also emphasized the importance of case coordinators in the management of referrals and transfers of care between different specialists in the MDT. These clinicians ensure that patients are not lost to follow-up between referrals. Specialist nurses such as diabetic, endocrine, and pediatric bone nurses were also highlighted as supporting case coordination and patient management in some centers.
Attendee quotation
• “Case coordinators are important in facilitating multi-disciplinary care by coordinating specialist clinicians in the MDT.”
Participants further underlined the role of social workers and charity coordinators in fostering relationships between patients, patient groups, XLH advocacy organizations, and non-governmental organizations. Patient-focused groups could also help organize social activities for patients and families affected by XLH.
Challenges When Establishing an MDT
Establishing an MDT can be difficult even in large institutions with numerous specialists (Box 4). The fundamental challenge lies in locating and assigning all the specialists necessary to optimize the management of XLH and finding those willing to collaborate in the treatment process. Furthermore, there is a paucity of clinical care coordinator roles in many GCC centers, resulting in specialist physicians taking on these functions. Centers of excellence or national referral centers could be established, where fully functioning MDTs treat XLH patients and act as points of reference for referrals and advisory bodies for clinicians outside the centers.
Challenges Associated with Establishing an MDT.

A well-integrated, multidisciplinary physician network is key to supporting smooth pediatric-to-adult care transitions and delivering optimal management for adult patients. Few participants at the XLH Matters meeting reported that there was a fully established MDT at their center. To address this, suggestions included regional and international collaboration with relevant experts from different centers across the world via online platforms.
Summary
XLH care can be improved by the development of MDTs, including a range of specialists based on patient age and clinical needs. Resource limitations of both specialists and MDT coordinators were acknowledged. The formation of city-based MDTs spanning multiple centers or the establishment of national “centers of excellence” acting as regional hubs for referral and clinical advice were suggested as potential regional solutions.
The Psychosocial Aspects of XLH: Patient & Family Perspectives
Key Psychosocial Factors in XLH Management
Psychological and psychosocial factors are critical components in the management of XLH patients. The multisystemic impact of the disease can result in a substantial burden for patients beyond the direct manifestation of symptoms. 40 As such, the health-related quality of life (HRQoL) for patients with XLH tends to be low, and patients may have impairments across social functioning and mental health. 7 Moreover, the occurrence of neurological (Chiari malformations, spinal stenosis, craniosynostosis, hearing loss), endocrine (secondary and tertiary hyperparathyroidism, obesity, nephrocalcinosis), and psychological comorbidities (Box 5) are often higher in XLH patients compared with the general population. 40 The number of patients with XLH is also found to be higher in areas of greater deprivation. 40
The key psychosocial challenges discussed during the meeting are summarized in Box 5.
Psychological and Psychosocial Burden Associated with XLH.

When patients with XLH do not live near treatment centers, there may be a risk of loss to follow-up. This may be even more pronounced in patients receiving oral phosphate and active vitamin D treatment, as the need for frequent travel to specialist centers adds to the existing burden or their disease, and the burden associated with supplemental therapy.4,7,19 Use of telemedicine and user-friendly phone apps for symptom reporting may help reduce distance-related barriers to care.
The functional limitations associated with XLH lead to reduced QoL 7 and impact pediatric, adolescent, and adult patients differentially. 42 Among pediatric patients, functional limitations comprise delayed walking, gait abnormalities, and the use of walking devices. 7 The ability to engage in physical activity with other children is an important aspect of development, and motor impairments limiting mobility of play and independence have a substantial psychosocial impact on affected children. 7 Among adult patients, progressive symptoms of XLH, such as restricted range of movement, osteomalacia, stiffness, and fatigue, are likely to impair their ability to study, work, exercise, socially interact, and perform basic activities of daily living, ultimately leading to impaired QoL.7,41 While the psychological burden of XLH evolves over the course of the disease, it has been suggested that the greatest impact takes place during the adolescent years (see Figure 3), during which patients become increasingly aware of the lifelong impact of their condition. 42
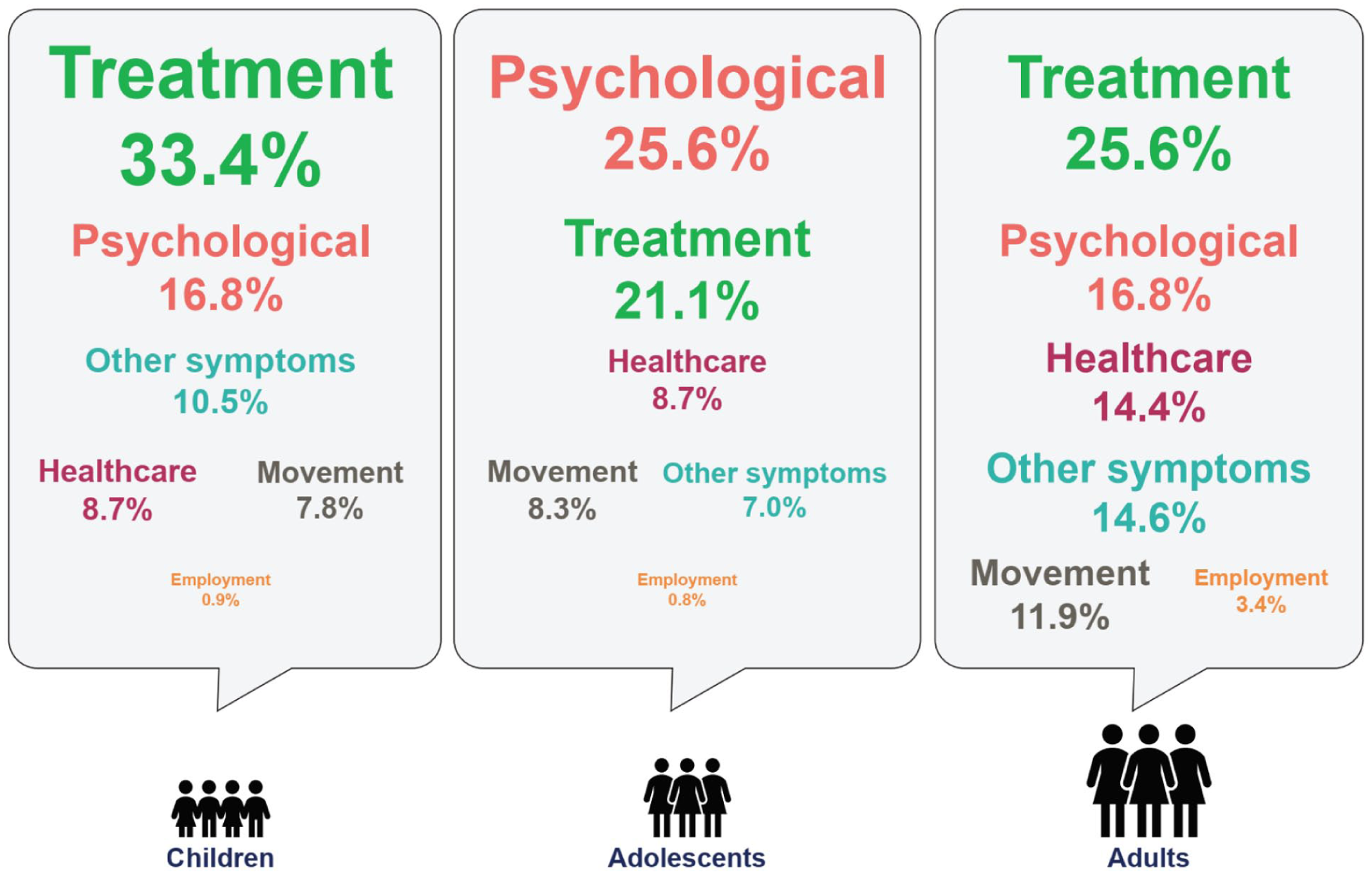
The evolving disease burden in patients with XLH, showing that psychological concerns manifest most among adolescents.
Evidence has demonstrated reduced HRQoL in XLH patients, with notably poorer scores on HRQoL metrics compared with the normative ranges. 7 The presence of comorbidities in XLH cases should also be acknowledged as a moderating factor on patients’ psychological health. The likelihood of mental health conditions, particularly depression, is found to be increased in XLH patients. 40
The Psychological Aspects of XLH: Patient and Family Perspective
Large and extended families are a distinctive feature of the population in the GCC, noticeably outnumbering those in Western nations. 21 Being an X-linked genetic disease, there are often multiple cases of XLH throughout the family tree (see Figure 4). 3

Family tree showing a typical affected family in the GCC area, illustrating how XLH can manifest across generations.
Attendee quotation
• “In the GCC we have big families, multiple marriages and multiple children. Affected children often grow up and have multiple children themselves.”
The psychological perception within families affected by XLH, both of the disease itself and of treatment, can have a significant influence across generations. People within these families may perceive XLH as a “normal” feature within their family and, therefore, lack understanding of the extent of the disease. The development of this perception of “normality” may be exacerbated by delayed diagnosis and insufficient supportive care for patients within affected families. Moreover, negative perceptions about the efficacy of oral phosphate and active vitamin D to treat XLH can also have profound impacts across generations, potentially discouraging families from seeking treatment due to a perceived lack of efficacy.
There is a need to raise awareness of the rarity and progressiveness of XLH among physicians and the public in the GCC. This would benefit families and increase the likelihood of timely diagnosis, ensuring effective treatment could be given throughout the family network after 1 member has been diagnosed. Further, a greater presence of patient advocacy groups and social support networks for patients affected by XLH would be useful in mitigating the psychological and psychosocial impact of the disease.
Summary
The psychological and psychosocial components of XLH patients are key considerations in the optimal management of the disease. Patients with XLH are likely to experience challenges beyond the direct symptomatic manifestation of their conditions. Mobility problems and pain can cause a range of downstream effects that negatively impact daily activities, education, social interaction, exercise, and work. XLH patients face an evolving burden of disease that is of greatest psychological impact in adolescence, during which patients may become particularly conscious of their conditions.
Few participants reported that psychological health was properly assessed among their patients, and the presence of psychological support teams was not widespread. A greater presence of support groups for patients with XLH is needed.
Addressing the Challenges of XLH Management
A contemporary review of the challenges of XLH management was provided in a presentation and open discussion session.
Physicians treating XLH are faced with 12 main challenges, summarized in Box 6.
Main Challenges Associated with XLH Management.

Challenge #1 in XLH management lies in the uncertainty of whether XLH is truly a rare disease, or if a portion of its low prevalence reflects underdiagnosis rather than epidemiological rarity. Rare diseases present numerous challenges to physicians, patients, and healthcare systems – 95% of rare diseases do not have specific treatments, and two-thirds of people living with rare diseases are children. 44 The estimated prevalence of XLH is 1:20,000-1:70,000, 45 though incidence rates may be affected by region, local population genetics, access to diagnosis and care, and underdiagnosis/misdiagnosis.18,46 Data from the XLH GCC Registry have revealed variability in the number of cases reported in each GCC region, implying underdiagnosis in some areas (assuming a roughly equal population distribution of XLH).31,47
Challenge #2 is the lack of awareness of XLH among physicians. In a survey of 68 fully qualified pediatric endocrinologists, only 26 (40%) were able to identify a hypothetical patient with classic signs of XLH. 43 Furthermore, 60% of physicians in the survey reported not testing for urine phosphate in cases of suspected HR. 43 In an effort to combat this, a detailed diagnostic algorithm for pediatric and adolescent patients with rickets and chronic low serum phosphate has been published for the GCC. 21 Greater dissemination of these resources among physicians is needed to improve awareness of XLH and the diagnostic resources available.
The phenotypic variation, nonspecific features, and overlapping clinical manifestations with other forms of rickets pose a further diagnostic Challenge (#3) in XLH care. One case study illustrated a patient with hallmark features of XLH, including hypophosphatemia, valgus deformity, sensorineural hearing impairment, and family history of HR. However, the etiology of the disease was ultimately revealed to be a novel pathogenic variant in the FOXI1 gene not recorded in the genetic database. 48 This case highlighted the importance of genetic testing as it directly influences treatment options; in this patient, burosumab would not be suitable despite the similar clinico-diagnostic features to XLH. As such, the nonspecific presentation of XLH creates a significant challenge in disease diagnosis and treatment decision-making.
Despite the importance of genetic testing in differentiating XLH from other forms of HR, 49 its widespread access is still lacking, which represents Challenge #4. The limited access to genetic testing also underlies other challenges, such as misdiagnosis and delayed diagnosis. 49 It was reported earlier in the pediatric workshop that access to genetic testing in the GCC remains variable due to either cost, availability, or waiting times for results.
Challenge #5 is delayed diagnosis. Even in straightforward cases of XLH with classic clinical symptoms, diagnosis may not be reached until a patient encounters a physician familiar with the disease. This is exacerbated by the fact that awareness of XLH remains low even among specialist physicians, 43 and timely diagnosis is not necessarily assured.
The need for surgery for severe deformities in XLH represents Challenge #6. Complex surgery may be required in cases of XLH which have progressed due to misdiagnosis or delayed diagnosis, with implications on healthcare costs and patient burden.
Challenge #7 involves cases of XLH which do not present in the bones. A case study of an 18-month-old male patient presenting with fascial cellulitis due to dental abscess was highlighted. The patient had low serum phosphate levels but no leg bowing or family history of consanguinity. However, the patient was born by cesarean section due to maternal bone deformities. Later analysis revealed a diagnosis of XLH, but the lack of clinical manifestations in the bones illustrates the potentially elusive presentation of XLH.
Challenge #8 is the frequent lack of detail in family history. Case study evidence was presented involving the mother of the aforementioned case. This patient had significant symptoms of XLH including bilateral leg deformities that required repeated corrective surgeries, as well as musculoskeletal pain and complete edentulism. The patient, however, was misdiagnosed with osteogenesis imperfecta and was not receiving any treatment. The significant symptoms of XLH in this patient should have been a clear indicator of the family history.
Challenge #9 comprises difficulties in the starting dose and subsequent titration of burosumab. Even when burosumab is administered according to standard dosing described in its Summary of Product Characteristics, 37 phosphate levels may not improve following treatment in certain cases. Decisions on future dose increase and titration can be challenging. Subsequent discussion clarified that burosumab treatment improves bone mineralization 32 and leads to a phenomenon in which the bone requires more calcium; this highlighted the need for patient monitoring that extends beyond phosphate levels alone, such as the monitoring of progress in physical functioning.
Challenge #10 involves the mode of administration and dosage preparations of burosumab. Currently, burosumab is available in preparations of 10, 20, and 30 mg/ml. 37 Administration can be challenging in patients where the dose involves less than a single vial, or where several vials are required. This can lead to drug waste in certain situations. Transitioning from traditional syringes to more dynamic iterations that permit flexible dosing could provide a potential solution to this challenge.
Challenge #11 is the lack of health equity/equality in XLH care. Finding treatment pathways for underprivileged patients is a difficulty that requires greater support involving patient advocacy groups.
Challenge #12 is the lack of evidence for treatments in cases of X-linked inherited hypophosphatemic rickets that are not XLH. Case study evidence was presented of an 18-year-old female patient with significant symptoms that had progressed through childhood. The patient has classic signs of XLH, but genetic diagnosis showed no pathogenic variant in the PHEX gene, and so the origin was autosomal dominant but not X-linked. Currently, there is insufficient clinical evidence to support the use of burosumab in this patient. More robust evidence is needed to support treatment decision-making in these patients.
Effective Transition From Pediatric to Adult Care – Challenges & Solutions
Transition Challenges in the GCC – Post-Adult Workshop Discussion
Following the earlier adult workshop, there was an open discussion among the attendees around the optimal age of transition of care in the GCC. Compared with the later transition ages in other countries, 9 this transition typically happens between the ages of 14 to 18 in the GCC, with attendees reporting challenges in transitioning patients at younger ages (Box 7).
Post-Adult Workshop Discussion – Transition at Age 14 is More Difficult Than at Age 18.

Determining the ideal transitional age for XLH patients is a complex clinical decision in which patient maturity, their level of understanding and readiness, available services, and funding of treatment must be considered. The education of general pediatricians and orthopedic surgeons is a foundational necessity that ensures effective referrals and coherent communication between pediatric and adult physicians. Currently, there is no standardized pathway for the transition of pediatric XLH patients to adult care in the GCC.
Effective Transition to Adult Care – Presentation and Discussion
The principles of effective transition were discussed in a presentation and open discussion. Transition from childhood to adulthood in XLH patients is characterized by an evolution of symptoms 50 and a change in their impact on patients’ lives. 42 The progressive nature of XLH means that symptoms presenting in adulthood are often developments of childhood symptoms, and poor management in childhood is correlated with worse outcomes in adulthood. 9
Attendee quotation
• “Adolescents have a combination of the symptoms seen in pediatric and adult patients, yet they often receive the least attention in social and medical care”
The adolescent years, marked by a combination of pediatric and adult symptoms (see Figure 5), 42 are often overlooked in patient management but represent an important period of development in the symptomatic burden of XLH. Adolescent XLH patients reported that psychological burden is of greatest impact, in contrast to pediatric and adult patients who reported that therapy is the most burdensome. 42 This illustrates how adolescent patients are insufficiently prepared to accept and take control of their condition to transition into adult care.
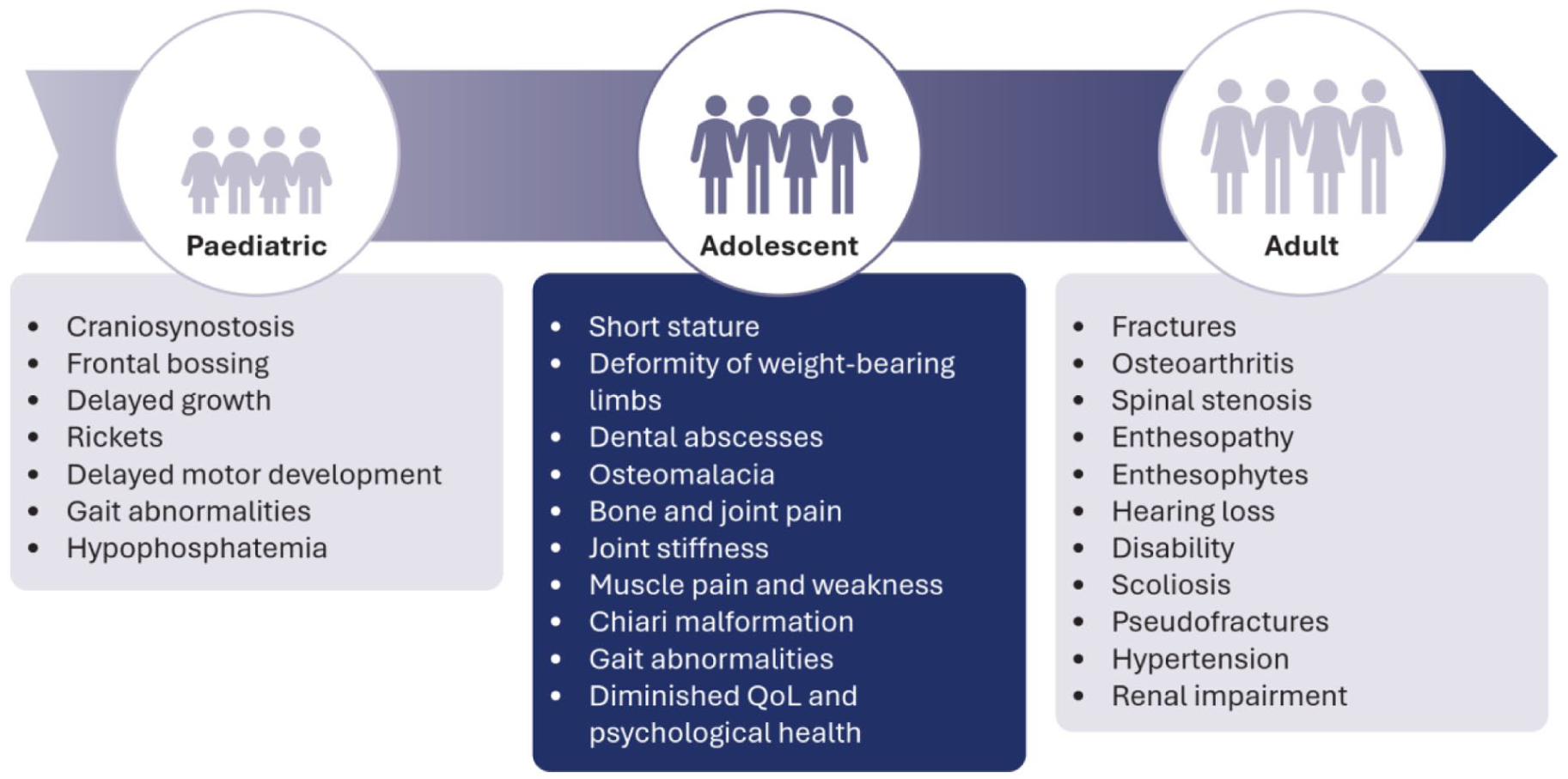
Attendee quotation
• “Adolescents report psychological impact as the major burden of XLH, reflecting how little they are prepared to deal with their disease and use health services effectively” (See Figure 3)
Principles of Effective Healthcare Transition and Potential Barriers
As with all chronic conditions, the transition from pediatric to adult care should be a structured process that systematically moves young patients with chronic conditions or disabilities from pediatric to adult healthcare services.9,51 This process should involve several intermediary stages and should not be seen as a simple shift to adult care.9,51
Attendee quotation
• “Transition from pediatric to adult care is a process, not a jump”
The major objective of pediatric-to-adult transition is to improve the ability of youth/young adults to manage their healthcare and independently utilize health services. A structured transition can lead to improvements in patient health, adherence to medication, and improved utilization of healthcare.52,53 Crucial to this is an organized process between pediatric and adult practices that facilitates patient–physician preparation, transfer of care, and integration into adult services. Achieving an effective transition is contingent on 6 core elements 54 and is subject to several barriers (summarized in Box 8).
The 6 Core Elements of Effective Transition of Care. 54

In addition to the lack of a widely adopted, standardized transition model in the GCC, misconceptions that XLH is only a childhood disease can lead to gaps in care during adolescence. 9 Disease management often stops when patients reach adolescence since standard clinical practice suggests discontinuing oral phosphate and active vitamin D once skeletal growth is complete. 9 Consequently, treatment may only resume when symptoms appear in adulthood. 9 Treatment gaps like this can lead to loss of follow-up and missed opportunities to improve health outcomes. 9
The variations in healthcare systems within the GCC also reflect differential priorities between pediatric and adult care. Pediatric care tends to be family-oriented, reliant on parental decision-making, and more focused on monitoring puberty and growth. Adult care, in contrast, tends to be patient-oriented, reliant on individual decision-making, and increasingly focused on fertility and family planning. Pediatric healthcare systems are also more likely to have MDT involvement than adult services.
Poor transition planning is often associated with worse outcomes for XLH patients. 9 Delays in medical care, loss to follow up, poor treatment compliance, and increased rates of hospital visits are all potential consequences of ineffective transition.
A Proposed Model for Effective Transition From Pediatric to Adult Care
An optimal transition from pediatric to adult healthcare should follow a structured educational timeline (see Figure 6). This involves a preparatory process that empowers patients with self-management skills appropriate to their age. A patient’s readiness for transition should be assessed with questionnaires to ensure they are well prepared to take responsibility for appointment keeping, medication management, and their own health status. 9
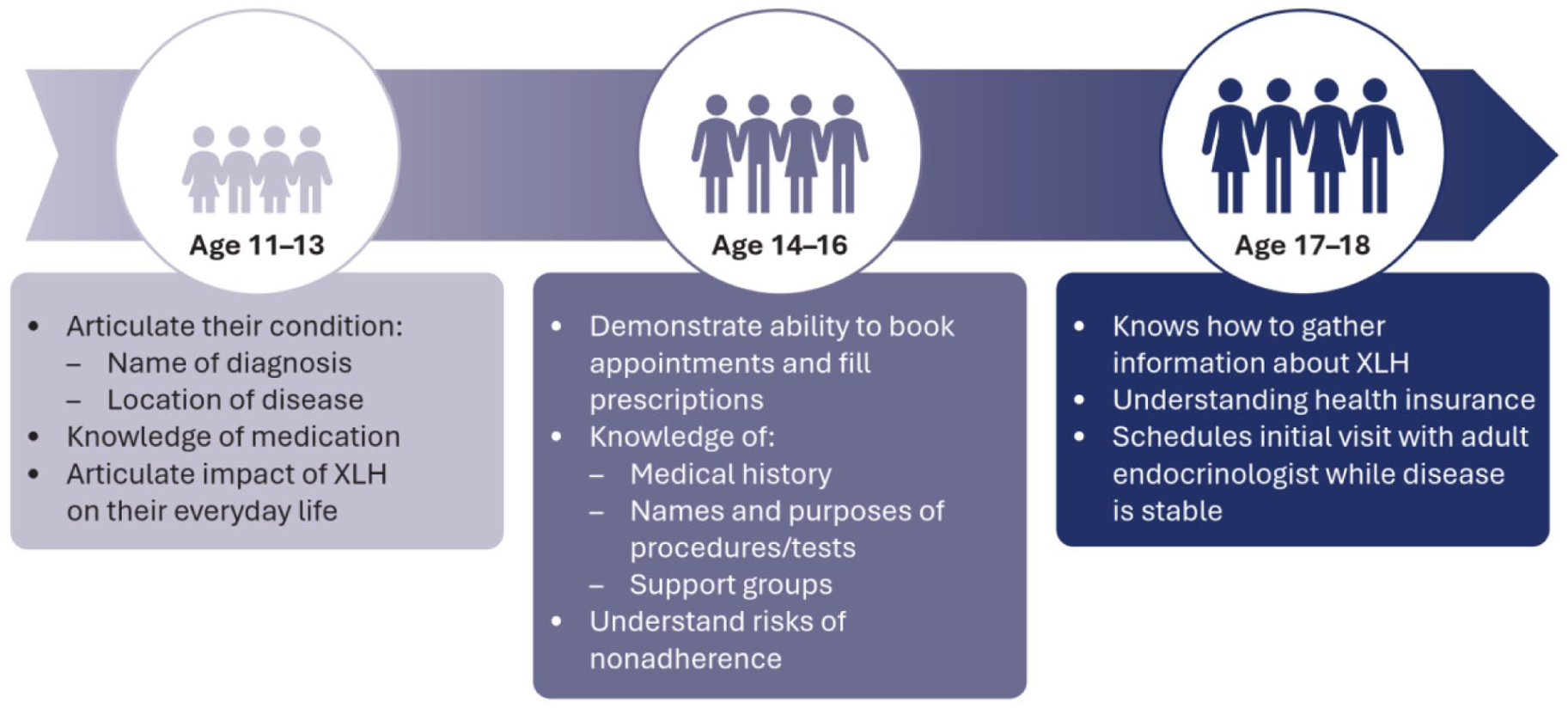
A timeline of patient empowerment underpins an effective transition from pediatric to adult care.
Several elements of effective transition have been implemented in individual centers, and these are summarized in Box 9. Expert consensus recommendations on transition of adolescent XLH patients to adult care have also been published. 9 However, at the time of writing, there is no globally standardized model for the transition of pediatric XLH patients to adult care.
Elements of Optimal Transition Established Locally.

An existing local transition model at the King Abdullah Specialized Children’s Hospital (KASCH), Saudi Arabia, uses an adolescent clinic to facilitate the transition process; this could act as a template for the wider GCC region. The transition model (Figure 7) distinguishes adolescent patients aged 14 to 18 as the key transitional group, with preparation starting before age 12.

Proposed model of transition illustrating the necessary preparations for transition and the dynamics of the transitional group.
Transition involves the development of patient decision-making and self-management skills, as well as providing access to support groups, educational materials, and community services. Patients undergoing transition should receive collaborative care from pediatric and adult healthcare providers within an MDT to facilitate optimal information sharing and ensure commitment of both services to continuing care for the patient.
During the transition process, involved physicians should hold MDT reviews to monitor progress and anticipate potential gaps in care. Patient education should be provided sequentially, reflecting the patients’ age and maturity, as presented in Figure 6. Patients and their families should be key partners in discussions around the transition to adult care. Such discussions should occur well in advance, and coordination with adult physicians should be established before the transition is finalized.
Summary
The transition of pediatric patients to adult care is not a single-step transfer but an extensive process involving education, physician collaboration, and care planning. A successful transition relies on close collaboration between pediatric and adult caregivers within a multidisciplinary context.
The XLH Matters participants proposed a transition model highlighting the adolescent years from 14 to 18 as the key period, which could act as a template for optimal transition across the GCC region. Local adaptation according to specialist availability and geographical location of centers is feasible but should be planned early in the patient’s lives and implemented based on the principles of good transition.
Updates to the International XLH Guidelines
At the time of XLH Matters 2024 GCC Edition, the latest XLH clinical guidelines 15 were unpublished. A summary preview of the key elements of the new guidelines was shared at the meeting, with additional discussion around the key points.
Guideline Recommendations on the Diagnosis of XLH
A summary of the key guideline recommendations for the diagnosis of XLH from the latest guidelines was presented, summarized in Box 10.
Recommendations for the Diagnosis of Children, Adolescents, and Adults with XLH.

The diagnostic workup for XLH patients should comprise a detailed family history and clinical evaluation assessing evidence of rickets, growth failure, and dental abnormalities (not only in cases of abscesses but as a standard approach). 15 Radiographic evaluation should be performed to grade the severity of rickets, as this helps to gauge the efficacy of treatment over time; healing rickets is key to confirming the benefit of treatment. 15
Biochemical testing should be performed as part of the workup, comprising fasting (in adults) serum phosphate levels, calcium, total or bone-specific (in adults) alkaline phosphatase (ALP), PTH, 25(OH) vitamin D, 1,25(OH)2 vitamin D, intact FGF23 (when available), and creatinine. Urinary levels of calcium, phosphate, and creatinine should also be assessed using a spot urine or 24-hour urine collection for the calculation of TmP/GFR and urinary calcium/creatinine ratio (in adults). 15 In children, collecting a 24-hour urine can be difficult, so spot urine can be used instead. The calculation of TmP/GFR and calcium/creatine ratio is a crucial step to inform the physician on calcium loss.
Ultimately, diagnosis in children and adults should be confirmed by genetic testing when possible. 15 If genetic analysis is unavailable, diagnosis should be supported by a positive family history and elevated FGF23 levels. 15
In cases where a family history of XLH is present, pre-implantation genetic testing for PHEX variants may be employed, provided it conforms to country-specific legal and ethical standards and is communicated through appropriate genetic counseling. 15 Genetic counseling is especially important to patients transitioning from pediatric to adult care as well as families affected by XLH who are planning pregnancies. 15
Follow-Up of Patients with XLH
Detailed recommendations for the follow-up schedule of pediatric, adolescent, and adult patients are provided in the recent XLH clinical guidelines. 15 Guidance is provided for the frequency of follow-up across the MDT. 15 Children and adolescents demonstrating satisfactory response to treatment should be evaluated at least every 6 months, while more regular follow-up (e.g. every 3 months) should occur in periods of rapid growth or at initiation/modification of therapy. 15
For adult patients no longer undergoing skeletal growth, assessments are conducted less frequently. Adults should be assessed every 3 to 6 months initially if receiving treatment, or every 6 to 12 months once stabilized. 15 Growth assessments may still be useful in adult patients, as height can decrease due to spinal deformities; this can be detected by height assessment. Objective measures of physical functioning among adults (e.g. the 6-minute walk test, Timed Up and Go [TUG] test) are also useful, as they may provide a better indication of symptom improvement than self-reports.55,56
In addition, assessment of possible treatment complications is essential and includes renal ultrasound to monitor potential nephrocalcinosis, regular radiographic assessments to grade the severity of rickets, regular dental follow-up, magnetic resonance imaging, and neurological follow-up to monitor potential craniosynostosis (particularly among adult patients). 15 Of note, attention to dental follow-up is important, since long durations between follow-ups can lead to dental deterioration that is difficult to remedy.
Treatment Recommendations for XLH
Burosumab is the recommended treatment for children and adolescents (aged 1-17 years) with an established XLH diagnosis and signs of rickets – including leg deformities, elevated total ALP, and/or radiological evidence of rickets. 15 Among asymptomatic adults, the routine treatment of XLH is not recommended; however, careful clinical assessment is needed to establish if patients are truly asymptomatic or if behavioral adaptation as a result of symptoms (e.g. minimizing activity) has occurred. 15 On the other hand, adult patients with significant symptoms of XLH should be treated. Burosumab is recommended in adult patients with pseudofractures or insufficient skeletal response to oral phosphate and active vitamin D; whereas oral phosphate and active vitamin D is recommended in adult patients with biochemical and/or clinical signs of osteomalacia, musculoskeletal pain, or stiffness. 15
In infants (aged <1 year) who are not eligible for burosumab treatment, as well as in children and adolescents (aged <18 years) where burosumab is not available, oral phosphate and active vitamin D is recommended once an XLH diagnosis is established. 15
It was noted that adding oral phosphate and active vitamin D to burosumab treatment is formally contraindicated. 15
Defining and Monitoring Response to Therapy
Defining treatment response is important when guiding decisions over future therapy.
A complete response to oral phosphate and active vitamin D is unlikely in patients with XLH. However, a satisfactory response may be defined in children as a significant improvement in rickets within 12 months, assessed by bone pain, serum ALP levels, and radiologically graded rickets severity. 15 In adults, satisfactory response is defined as a significant improvement in musculoskeletal pain, stiffness, and osteomalacia, as well as normalization of ALP levels (if elevated) within 24 months.
A satisfactory response to burosumab in children comprises significant improvements in age-adjusted serum phosphate levels and rickets severity, assessed by bone pain and serum ALP, within 6 months. Within 24 months, response is defined by improvements in leg deformities, serum ALP values, and normal growth velocity (>25th percentile for sex and age). 15 In adults, treatment response is defined by significant improvements in renal phosphate wasting, serum phosphate levels, and musculoskeletal pain within 6 months 15 ; within 12 months, there should be improvements in musculoskeletal pain, stiffness, osteomalacia, and total or bone-specific ALP. 15
Summary of Treatment Considerations
Treatment started in childhood should be continued across the transition period into adulthood. Ongoing treatment is needed because growth has not finished at the time of growth plate fusion, and bone mass may be optimized by therapy. 15 Current guidelines recommend continued treatment with burosumab into the third decade of life (the time of peak bone mass). 15 In cases where burosumab is not available, oral phosphate and active vitamin D are recommended. 15
Treatment with burosumab should be discontinued in patients considering pregnancy. 15 In these patients, treatment with oral phosphate and active vitamin D can be considered in the last trimester of pregnancy. 15
There appears to be a subset of patients who will not reach normal serum phosphate levels following treatment with burosumab, despite improvements from baseline. 57 Nonetheless, these patients may still attain clinical benefits in physical functioning, pain, and musculoskeletal performance with burosumab treatment. 57
Concluding Remarks
The XLH Matters 2024 GCC Edition was a first-in-region networking and educational meeting held in the GCC to encourage the sharing of real-world data and clinical recommendations and to improve the management of patients with XLH in the Gulf region. Attending expert clinicians shared updates on best practices in identifying XLH in pediatric and adult patients and the optimal management of XLH across the lifespan. Participants also exchanged insights regarding the challenges in XLH care, transition from pediatric to adult care, collaboration within an MDT, and the psychosocial impacts of XLH.
Increased education and awareness in XLH among primary physicians is key to ensuring timely diagnosis and access to proper treatment. The unique, extended family structures commonly found in the GCC mean that cases of XLH may arise throughout the family tree. Hence, obtaining a detailed family history is important for diagnosis, while pedigree analysis may help identify undiagnosed family members.
A progressive, collaborative model for transitioning pediatric patients into adult care proposed to ensure continuity of care.
Overall, attendees of XLH Matters 2024 GCC Edition agree that patient care across the lifespan can be enhanced in the GCC region by sharing knowledge, best practices, research findings, and clinician collaboration.
Footnotes
Acknowledgements
The authors wish to thank Highfield Communication Ltd for provision of medical writing assistance, which was funded by Kyowa Kirin.
Ethical Considerations
The authors confirm that no ethical approval was required for this manuscript as no human participants or scientific data are reported.
Consent to participate
The authors confirm that no consent to participate was required for this manuscript as no human participants were involved.
Consent for Publication
All authors provide their consent for publication in the journal Therapeutic Advances in Endocrinology and Metabolism.
Author Contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This supplement was based on the XLH Matters 2024 GCC Edition meeting which was organized and sponsored by Kyowa Kirin. Medical writing support was funded by Kyowa Kirin.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors received an honorarium from Kyowa Kirin to present at the XLH Matters 2024 GCC Edition meeting. Financial support for accommodation during the meeting was provided by Kyowa Kirin; authors from outside the UAE received support from Kyowa Kirin to travel to the meeting. LS has received grants/contracts from Alexion/AstraZeneca, Chiesi and Kyowa Kirin. He has also received honoraria for lectures and advice from Alexion/AstraZeneca, AM-Pharma, Amgen, Alesta, BioMarin, Chiesi, Gedeon-Richter, Haleon/GSK, Inozyme, Ipsen, Kyowa Kirin, Mereo, Novartis, Stadapharm, Theramex, UCB and Ultragenyx. MAB has no additional conflicts of interest. HF has no additional conflicts of interest. Dr Al-Sagheir has received honoraria for medical writing from Kyowa Kirin. TMF has no additional conflicts of interest. AD has no additional conflicts of interest. Dr Sumaily has no additional conflicts of interest. NA has no additional conflicts of interest. AH has received consulting fees from Kyowa Kirin. He has also received speakers’ honoraria from Kyowa Kirin. He has also participated in advisory boards for Kyowa Kirin. AA has no additional conflicts of interest. MZM has received support for attending meetings/travel grant from BioMarin for attending the 62nd Annual ESPE conference, 2024.
Data Availability Statement
The authors confirm that the findings and content of this supplement are available within the article.
Glossary

| Term | Definition |
|---|---|
| 1,25(OH)2D (Calcitriol) | The biologically active form of Vitamin D is produced in the kidneys. It binds to the Vitamin D Receptor (VDR) to regulate calcium and phosphorus levels. |
| 25(OH)D (25-hydroxyvitamin D) | Primary circulating form of vitamin D and the most reliable biochemical marker for assessing vitamin D status; low levels support a diagnosis of calcipenic rickets. |
| Autosomal X-linked recessive disease (e.g. Dent’s or Lowe’s disease) | Rare hereditary renal tubular disorders that cause phosphate wasting and can produce hypophosphatemic rickets; they mimic XLH biochemically and require family history and/or genetic testing for differentiation. |
| Blount’s disease | A growth disorder of the medial proximal tibial physis leading to progressive genu varum (bowing). |
| Calcification of ligaments and joint capsules | Abnormal mineral deposition around joints, often seen in older adolescents and adults with XLH. |
| Calcipenic rickets | Rickets caused by calcium or vitamin D deficiency, typically associated with elevated PTH and distinct from phosphopenic rickets such as XLH. |
| Chiari malformation | Structural defects in which part of the brain’s cerebellum extends downward through the foramen magnum into the spinal canal, potentially causing headaches, balance issues, or neurological symptoms. |
| Craniosynostosis | Premature fusion of one or more cranial sutures, altering skull growth and potentially increasing intracranial pressure or causing head-shape abnormalities. |
| Cupping, metaphyseal | Radiographic sign in rickets where the metaphysis appears concave due to impaired mineralization. |
| Dental abscesses | Pulp infections developing without caries or trauma, common in XLH due to defective dentine and enamel mineralization. |
| Dental enamel hypoplasia | Underdeveloped or thin enamel, commonly seen in nutritional rickets and distinguishable from the dentine-related defects typical of XLH. |
| Endocrinologist | Specialist who manages metabolic bone disorders, including XLH, overseeing phosphate therapy, burosumab, and long-term endocrine complications. |
| Enthesopathy | Pathologic involvement of tendon or ligament insertions into bone, often presenting as pain or stiffness and common in adult XLH. |
| ENT (ear, nose, and throat) specialist | Clinician managing ear, nose, and throat complications such as conductive or sensorineural hearing loss that can occur in adults with XLH. |
| Exome | The portion of the genome consisting of all protein-coding regions (exons) of genes. Although exons represent only ~1% to 2% of the genome, they contain the majority of known disease-causing genetic variants. |
| Genu valgum | Medial angulation of the knees (“knock-knee” deformity), frequently observed in pediatric XLH. |
| Genu varum | Lateral bowing of the legs (“bow-legged” appearance), a classic sign of rickets seen in XLH. |
| Hypercalciuria | Excess calcium excretion in urine, often associated with prolonged treatment using oral phosphate and active vitamin D analogs. |
| Metaphyseal fraying | Radiographic irregularity of the metaphyseal margin due to defective mineralization, characteristic of rickets and XLH. |
| Metaphyseal widening | Expansion of the metaphysis seen in pediatric rickets secondary to disordered mineralization. |
| Nephrocalcinosis | Deposition of calcium salts in renal tissue, associated with long-term oral phosphate and active vitamin D therapy. |
| Neurologist | Specialist who may be involved in XLH care when neurological manifestations occur, such as craniosynostosis-related symptoms or neuropathic complications. |
| Neurosurgeon | Consulted in XLH cases with craniosynostosis, Chiari malformation, or severe spinal stenosis requiring operative intervention. |
| Obesity | A chronic condition characterized by excessive accumulation of body fat and associated with increased cardiometabolic and mechanical health risks. Common in patients with XLH due to reduced physical secondary to pain. |
| Obstetrician/gynecologist | Manages reproductive and pregnancy-related considerations in individuals with XLH, including fertility, pregnancy planning, and decisions around burosumab use. |
| Osteoarthritis | Degenerative joint disease common in adults with XLH due to chronic abnormal loading, malalignment, and altered bone mineralization. |
| Osteomalacia | Impaired bone mineralization in adults leading to soft bones, bone pain, pseudofractures, and characteristic radiographic findings. |
| Osteophyte formation | Growth of bony outcrops along joint margins, frequently observed in adult XLH as a component of degenerative joint changes. |
| Orthopedic surgeon | Medical specialist managing skeletal abnormalities such as limb bowing, pseudofractures. May perform corrective surgery in moderate to severe XLH. |
| Parathyroid hormone (PTH) | Hormone that regulates calcium and phosphate homeostasis; elevated in calcipenic rickets and helps differentiate rickets types. |
| Pediatric endocrinologist | Leads diagnosis and treatment of XLH in children, including growth monitoring, biochemical evaluation, management of rickets, and genetic confirmation. |
| Phosphopenic rickets | Rickets caused by inadequate phosphate availability, characterized by low serum phosphate and typically normal or mildly elevated PTH. |
| Physical therapist | Supports functional mobility, strengthening, gait training, and pain management in both pediatric and adult XLH populations. |
| Psychiatrist | Treats mental health conditions such as depression and anxiety, which can be more common in those with chronic XLH. |
| Psychologist | Provides behavioral and emotional support to address the psychosocial burden of chronic disease in children and adults with XLH. |
| Pseudofractures (looser zones) | Incomplete fractures resulting from under mineralized bone; hallmark radiologic feature in adult XLH, often responding well to burosumab therapy. |
| Renal phosphate wastage | Loss of phosphate through the kidneys due to elevated FGF23 or other renal tubular defects, resulting in chronic hypophosphatemia. |
| Rheumatologist | Medical specialist managing chronic musculoskeletal pain, stiffness, osteoarthritis, and enthesopathy commonly seen in adults with XLH. |
| Schmid metaphyseal chondrodysplasia | Genetic skeletal disorder causing metaphyseal changes and limb deformities similar to XLH, making it an important differential diagnosis for pediatric rickets. |
| Secondary Fanconi syndrome | An acquired generalized proximal tubule dysfunction causing phosphaturia and other solute losses, which can mimic XLH biochemically. |
| Secondary hyperparathyroidism | Compensatory overproduction of PTH in response to chronic hypocalcemia or abnormal mineral metabolism, often seen in chronic phosphate-wasting conditions or vitamin D deficiency. |
| Sensorineural hearing loss | Hearing impairment caused by dysfunction of the inner ear (cochlea) or auditory nerve pathways, commonly presenting with reduced sound clarity or difficulty distinguishing speech. |
| Skeletal dysplasia | Broad category of genetic bone-growth disorders; some produce radiologic features that overlap with those of XLH. |
| Sclerosis | Increased radiodensity of trabecular bone at distal long-bone ends, seen in older adolescents and adults with XLH. |
| Spinal stenosis | Narrowing of the spinal canal due to osteophytes, ligamentous calcification, or skeletal abnormalities occurring in adult XLH. |
| Tertiary hyperparathyroidism | Autonomous PTH overproduction following long-standing secondary hyperparathyroidism, such as during chronic oral phosphate therapy. |
| Whole exome sequencing (WES) | A type of genetic analysis that sequences only the exons (the protein-coding regions) and is more accessible and affordable than WGS. |
| Whole genome sequencing (WGS) | A type of genetic analysis that sequences both protein-coding regions and non-coding regions (often called “junk DNA”), which are increasingly recognized for their role in regulating gene expression. More costly than WES. |