
Abstract
Clitoromegaly in a newborn with otherwise typically appearing female genitalia indicates exposure to excess androgens in fetal life. Congenital Adrenal Hyperplasia (CAH) is the most common cause of virilization at birth and empiric treatment is often pursued due to the risk of life-threatening adrenal crisis. Turner syndrome (TS) does not usually present with atypical genital appearance or other signs of hyperandrogenism, unless Y chromosome material is present. In this report, we describe a newborn who was noted to have clitoromegaly soon after birth with an otherwise normal exam and no syndromic features. Empiric treatment with hydrocortisone and fludrocortisone was started while initial laboratory results were pending. However, prior to receiving CAH hormonal panel results, karyotype testing returned as [45,XO(3)/46,XX(17)], consistent with mosaic TS. Testing for Y chromosome material, pursued as a potential cause of clitoromegaly, was negative. To add to the diagnostic dilemma, newborn screening for CAH was also negative. Results from the CAH panel done soon after birth eventually returned and revealed an elevated 17-hydroxyprogesterone level (17-OHP) of 1540 ng/dl, which is more congruent with non-classical CAH and does not typically result in clitoromegaly at birth. To clarify the diagnosis, a 250 mcg cosyntropin stimulation test was performed after holding steroids for a week. Post-stimulation 17-OHP of 82334 ng/dL confirmed classical CAH secondary to 21-hydroxylase deficiency and genetic analysis identified biallelic pathogenic deletions of CYP21A2 gene. Current TS guidelines recommend testing for Y chromosome material if masculinizing features develop. This case underlines the value of including CAH in the differential diagnosis as another potential cause of virilization in patients with TS. The concurrence of CAH and TS poses added complexity for clinical management due to their overlapping impacts on linear growth, puberty, reproductive function, bone density and metabolic health.
Plain Language Summary
This report describes a newborn girl who had an enlarged clitoris at birth, which can be a sign of exposure to high levels of male hormones before birth. The most common cause of this is Congenital Adrenal Hyperplasia (CAH), where the adrenal glands cannot make enough cortisol and instead produce excess male hormones. Absence of cortisol can result in a life-threatening condition called adrenal or salt wasting crisis, so treatment for suspected CAH is often started right away while awaiting test results. While CAH testing results were still pending, genetic testing revealed the baby had Turner Syndrome (TS), a condition where girls are missing all or part of one X chromosome. TS does not usually cause masculinization unless they also have Y chromosome material in their cells, but testing for Y material was negative in this baby. Adding to the puzzle, the newborn screening test for CAH also came back negative. However, more detailed blood tests, including a “stimulation test” showed elevated hormone levels consistent with CAH due to 21-hydroxylase deficiency, the most common enzyme deficiency causing CAH. Genetic testing found that both copies of the responsible gene had deleted sections. This is a rare case of a baby having both TS and CAH at the same time. Current guidelines recommend testing for Y chromosome material when girls with TS show masculine features. This case suggests that CAH should also be considered as a potential cause of hyperandrogenism. Managing both conditions together can be challenging because they both affect growth, puberty, fertility, bone health and metabolism.
Keywords
Introduction
Differential Diagnoses for Congenital Clitoromegaly

Abbreviation: DSD: Disorder of Sexual Development.
Turner Syndrome (TS) is a sex chromosome disorder that affects phenotypic females and is caused by the complete or partial loss of an X chromosome. Around 40-50% of those with TS have 45, X karyotype (monosomy X) and the rest have varying forms of mosaicism where there is a normal cell line and an abnormal second (or third) cell line (45,X/46,XX or 45,X/47,XXX or 45,X/46,XY etc). X chromosome anomalies such as partial deletions, ring chromosome X or isochromosome Xq can also occur. Compared to those with 45,X karyotype, those with mosaicism tend to have a milder phenotype although this can vary considerably based on the degree of mosaicism and body tissues affected. Around 10-12% of those with TS have mosaicism that involves a cell line containing Y chromosome material. 5 These individuals can have ovotesticular/testicular tissue in their dysgenetic gonads and may present with signs of hyperandrogenization such as clitoromegaly, premature pubarche, hirsutism or deepening of voice. An increased prevalence of germ cell tumors (gonadoblastoma, dysgerminoma) has been noted in TS individuals who carry Y chromosome material and current guidelines recommend considering prophylactic gonadectomy in this situation.5,6 The presence of Y chromosome material may be identified on a standard karyotype however, use of FISH, real time PCR or whole-exome sequencing are more sensitive in detecting cryptic Y material. 7
In this report we describe a case of congenital clitoromegaly where establishing the cause proved challenging as the patient’s two co-existing conditions that could independently cause fetal hyperandrogenemia and clitoromegaly.
Case Description
A term infant born at 37 weeks gestational age via cesarean section to non-consanguineous parents was noted to have clitoromegaly soon after birth. The 33-year-old G3P2 mother had a pregnancy that was complicated by gestational hypertension and hypothyroidism, which was appropriately treated with levothyroxine. There was no known family history of virilization in women, infertility or sudden infant death. Birth weight was 3.29 kg and birth length was 52 cm. Genital exam revealed clitoromegaly (length 1.5 cm, width 1 cm) with separate urethral meatus and vaginal openings and no palpable gonads (Prader Stage II). The rest of the physical exam was normal except for mild respiratory distress. No dysmorphic features were noted. The patient was transferred to the neonatal ICU due to respiratory distress requiring CPAP support and high clinical suspicion for CAH, the most common cause of virilization at birth. Initial labs were sent on the day of birth including electrolytes, karyotype and CAH profile 6 (a comprehensive adrenal hormone panel that screens for different forms of CAH). Patient was then empirically started on hydrocortisone 2.5 mg three times a day (30 mg/m2/day) and fludrocortisone 0.1 mg twice daily. Pelvic ultrasound revealed a uterus and bilateral fallopian tubes but was unable to visualize the ovaries. Respiratory distress noted after birth was transient, and she was weaned from CPAP to room air within a day. The state newborn screen (NBS) was sent at around 48 hours of life.
Prior to receiving results of CAH testing, karyotype resulted as [45,XO(3)/46,XX(17)], consistent with mosaic TS. This was surprising because TS does not usually cause virilization or present with atypical genitalia, except in the presence of Y chromosome material. To evaluate for Y chromosome material, fluorescence in situ hybridization (FISH) for sex-determining region Y (SRY) gene and Y centromere were sent. Given the diagnosis of TS, renal and cardiac imaging were obtained to screen for comorbidities. Renal ultrasound was normal. Echocardiogram showed a small atrial septal defect, mildly dilated ascending aorta and trivial mitral regurgitation. Blood pressure readings, blood glucose and electrolytes remained within normal limits throughout the admission, and the newborn tolerated oral feeds and gained weight appropriately. On day six of life, the patient was discharged home on steroid supplementation with cardiology and endocrinology follow-ups scheduled. At the time of discharge, the CAH profile 6, NBS, and FISH for SRY and Y centromere were still pending.
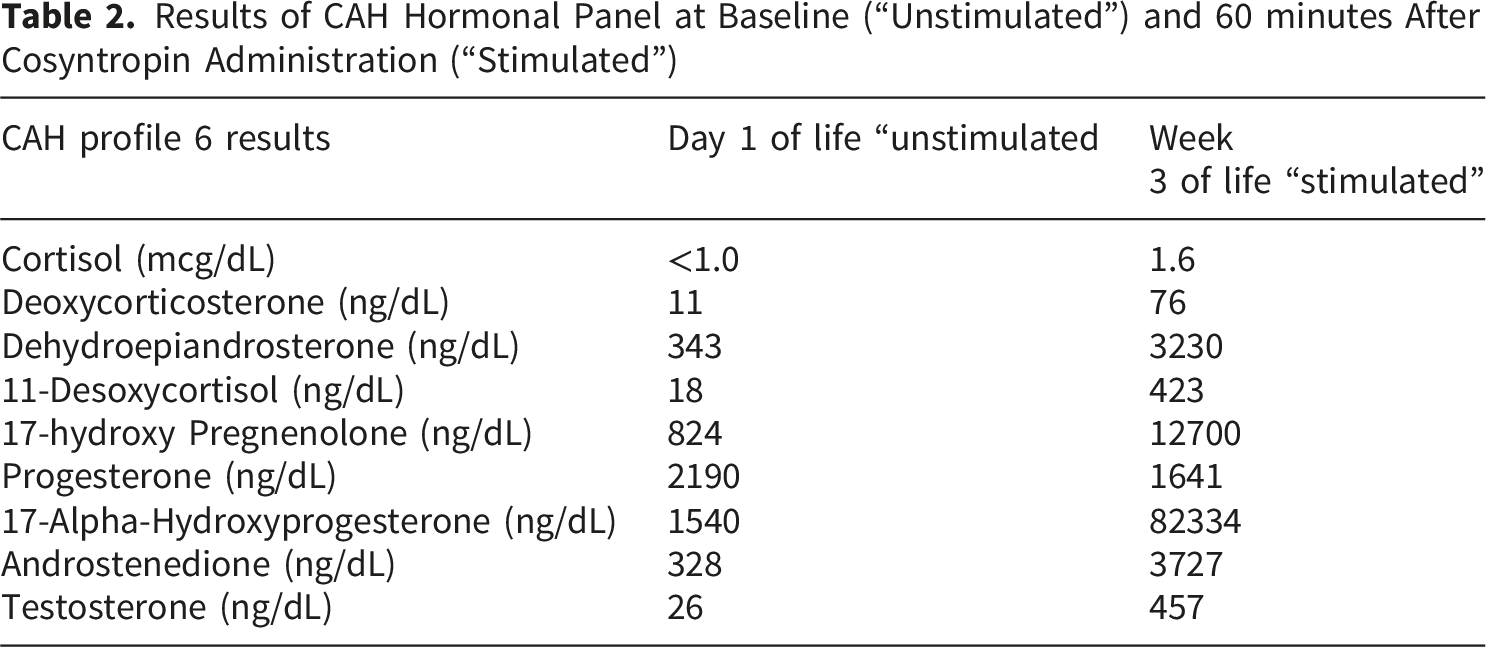
Over the next two weeks, pending labs resulted in the following order: 1.) FISH did not identify the SRY gene or Y centromere material, eliminating a possible cause of clitoromegaly, 2.) NBS was negative for CAH, however this result was unreliable as it was collected after initiation of steroids which can suppress the adrenal axis and lead to a false negative screen, 3.) Lastly, the CAH panel sent on the day of birth showed an elevated level of 17-alpha-hydroxyprogesterone (17-OHP) at 1540 ng/dl. However, this level was not as high as that typically seen in patients with classical CAH presenting with clitoromegaly. 8
Results of CAH Hormonal Panel at Baseline (“Unstimulated”) and 60 minutes After Cosyntropin Administration (“Stimulated”)
Trends in CAH Lab Profile and Hydrocortisone Dosing Over Time
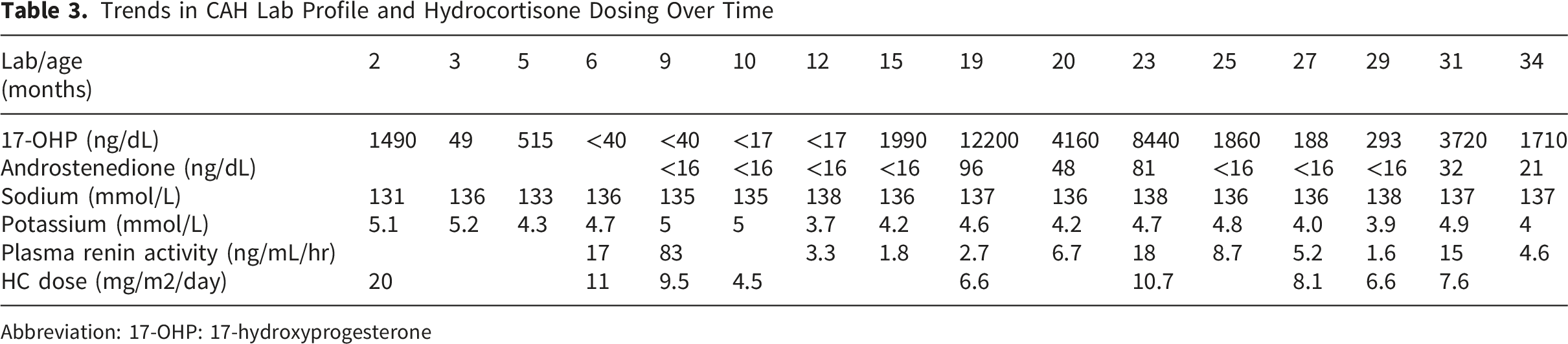
Abbreviation: 17-OHP: 17-hydroxyprogesterone
Discussion
This infant was diagnosed with concurrent mosaic TS and CAH due to 21-hydroxylase deficiency. Establishing the cause of clitoromegaly proved challenging since both these diagnoses could potentially cause hyperandrogenemia. The timeline of when the labs resulted, false negative NBS for CAH, the mild phenotype associated with mosaic TS and lower than expected baseline 17-OHP level added to the complexity of the case. CAH is the most common cause of virilization at birth and infants are often empirically treated for it until the diagnosis can be clarified. Virilizing signs in patients with TS are unusual but can occur in the presence of a Y chromosome bearing cell line. However, in our patient no Y chromosome material was detected and the cosyntropin stimulation test and CYP21A2 gene testing were consistent with classical CAH, making CAH the likely cause of her clitoromegaly.
Hydrocortisone needs were transiently as low as 4.5 mg/m2/day between 10 and 18 months of age. The cause remains unclear. Although lower hydrocortisone requirements have been described in a subgroup of patients with classical CAH, they usually require a dose higher than 5 mg/m2/day. 9 A possible explanation is the inconsistent dosing provided by pharmacy-contained hydrocortisone suspensions, which the patient was receiving.
At the time of this publication there are 13 prior cases10-22 in literature that describe concurrent TS and CAH. Seven of the cases presented in the neonatal period with atypical genitalia and varying degrees of virilization,13,15-18,20 21 while other cases presented with signs of hyperandrogenism later in childhood10,12 or were diagnosed as adults.11,14,19 Thus, clinical presentation of concurrent TS and CAH can vary considerably. Most of these cases had mosaic TS and did not display obvious syndromic features as often seen with mosaicism. This means that the TS diagnosis may be missed entirely unless karyotyping is performed for another indication. The Inacio et al 22 case exemplifies this. They reported a patient who was prenatally diagnosed with mosaic TS (45,X/47,XXX) and had no phenotypic TS features but presented at 5 years of age with premature pubarche that led to the concurrent diagnosis of Non-Classical Congenital Adrenal Hyperplasia (NCCAH). Given that a karyotype is typically not included in the workup of premature pubarche, the diagnosis of TS may have never been made if not for prenatal testing. The finding that most of the concurrent CAH and TS cases involved mosaic TS karyotypes rather than 45,X monosomy is also consistent with the broader observation that approximately 80% of dual-diagnosis TS cases involve mosaicism, approximately twice the frequency in the general TS population. 23
Whether the co-occurrence of CAH and TS reflects a true association or is purely coincidental remains unclear, as studies assessing 21-hydroxylase activity in patients with TS have yielded conflicting results. Larizza et al 12 demonstrated that 21.5% of patients with TS had stimulated 17-OHP levels comparable to heterozygous carriers of 21-hydroxylase mutations, though this was not confirmed by gene analysis. Mantovani et al 24 similarly observed elevated post-stimulation 17-OHP levels in 35.4% of patients with TS. CYP21A2 gene analysis identified heterozygous mutations in 12.5% of the TS cohort, significantly higher than the 4.6% mutation rate in controls. Although these studies implicate a higher incidence of heterozygous carrier state for 21-hydroxylase deficiency in patients with TS, a later analysis by Onder et al contradicted this finding. 25 Only one of 44 subjects with TS had an elevated 17-OHP level in the carrier range, with no CYP21A2 mutation identified on gene analysis. Further research with larger sample sizes and systematic CYP21A2 genotyping across diverse populations is needed to better understand the potential relationship between CAH and TS.
Concurrence of CAH and TS poses added complexity for both its diagnosis as described in this patient as well as future clinical management. Short stature is the most common feature associated with TS and guidelines recommend initiation of growth hormone (GH) therapy when poor growth is noticed, as early as two years of age. 5 CAH also predisposes patients to short stature. Treatment of CAH is a balancing act between hyperandrogenism and hypercortisolism. Over treatment with glucocorticoids suppresses linear growth and undertreatment leads to adrenal androgen excess causing acceleration of growth, advancement of bone age, precocious puberty and premature fusion of the growth plates leading to compromised adult height. 2 Concurrent CAH and TS diagnosis will likely have a compound effect on the patient’s growth that requires careful monitoring and timely intervention. In addition to growth, concurrent TS and CAH may also have a dual impact on pubertal progression, reproductive function, bone density and metabolic health. Excess androgen production in inadequately treated CAH can hinder the hypothalamic-pituitary-ovarian axis, leading to anovulation, irregular menstruation and infertility. Patients with concurrent CAH and TS can experience delayed or stalled puberty, amenorrhea and infertility due to premature ovarian insufficiency even with proper management of CAH. Hormone replacement therapy may be required to induce puberty and maintain sexual development. The risk of low bone mineral density and fractures is increased in girls with TS due to estrogen deficiency as well as intrinsic bone abnormalities. Patients with TS also have significantly higher rates of central obesity, insulin resistance, type 2 diabetes, hypertension and dyslipidemia. Glucocorticoid therapy in CAH can further increase this risk for osteoporosis and poor metabolic health.
Conclusion
Congenital clitoromegaly is most often caused by CAH secondary to 21-hydroxylase deficiency and should be the presumed diagnosis until proven otherwise. Patients with TS do not develop virilizing signs, such as clitoromegaly, unless there is cryptic Y chromosome material that leads to gonadal dysgenesis and androgen production from testicular tissue in the gonads. This case underlines the value of including CAH in the differential diagnosis as another potential cause of virilization in patients with TS, particularly in the absence of Y chromosome sequences. The case also highlights that newborn screening for CAH can sometimes report false negative results and that patients with mosaic TS can have a very mild phenotype that can make the diagnosis challenging. The concurrence of CAH and TS in this patient poses added complexity for future clinical management due to their overlapping impacts on linear growth, puberty, reproductive function, bone density and metabolic health.
Footnotes
Ethical Considerations
Our study did not require an ethical board approval as written consent for the case report was provided by the patient’s legal guardian and no protected health information is included in this case report.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Prior Publication
Portions of this case were previously presented as an abstract at the Endocrine Society Annual Meeting, held in July 2025, and published in the Journal of the Endocrine Society, Volume 9, Issue Supplement_1, October-November 2025. This full case report provides detailed clinical analysis and additional data beyond the initial abstract.