
Abstract

Rosai-Dorfman disease (RDD), first described by Rosai and Dorfman in 1969, is a rare benign self-limited condition characterized by a non-Langerhans reactive histiocytic proliferation. 1 Clinically, it is characterized by a systemic involvement with high temperature, leukocytosis with neutrophilia, increased acute phase reactants, and polyclonal hypergammaglobulinemia, together with large, painless lateral-cervical lymphadenopathy, although occasionally other lymph nodes can be involved. 2 In about 40% of cases, there is extranodal involvement, the skin being the most frequently involved organ, followed by subcutaneous tissue, orbits, bones, and central nervous system, although theoretically any of the organs of the body economy can be affected. Very rarely, in about 3% of cases, the disorder is limited to the skin: this is cutaneous RDD (CRDD). 3 However, given the epidemiological data and the different clinical symptoms, some authors suggest that they are 2 different disorders 4 : RDD has an earlier onset (around the first and second decades of life), is slightly more predominant in black and white males and very rarely in Asian males, and has a variable prognosis. CRDD is more predominant in middle-aged women, has a high prevalence in Asian populations compared to RDD, and has a good prognosis.
Although the etiology is unknown in both conditions, hypotheses suggest the participation of some viruses (human herpesvirus 6, varicella-zoster virus, Epstein-Barr virus, and parvovirus B19) associated with some genetic factors that, in some cases, seem to be related to alterations in cell-mediated immunity. On the other hand, the polyclonal nature of this disorder would go in favor of a reactive inflammatory process rather than a neoplastic one. 5
Although the ways that CRDD presents are varied, its usual symptoms are papules, nodules, or painless reddish patches, either singular or multiple, on the face, torso, and/or limbs. Acneiform, pustular, and vasculitic presentations 5 ; lesions similar to annular giant granuloma 6 ; penile lesions as in Peyronie disease; cutaneous fistulas; ulcerated patches 7 ; and so forth have been reported.
Histologically, it is characterized by a dense diffuse inflammatory infiltrate of histiocytes occupying the dermis and sometimes the subcutaneous cellular tissue, together with lymphocytes, abundant plasma cells, and some neutrophils and eosinophils, usually adopting a nodular pattern and rarely a lobular one. Distinctively, the epidermis remains undamaged, and it rarely appears ulcerated. Histiocytoid cells, also known as Rosai-Dorfman cells, are large cells with soft and poorly defined edges, abundant clear eosinophilic cytoplasm, and a central nucleus with vesicular chromatin and prominent nucleolus. Emperipolesis is a very characteristic and important phenomenon for diagnosis. It involves the presence of inflammatory cells and/or undamaged erythrocytes contained in intracytoplasmic vacuoles, which, therefore, are protected from the cytolytic process deriving from common phagocytosis. Histiocytes are positive for S100 and CD68 and negative for CD1a, which, together with the absence of Birbeck granules (langerin negativity), allow for the ruling out of other conditions and confirm the diagnosis.5,6 Another characteristic finding is the presence of a fibrous tissue made up of fibrocollagen bundles, which intermingles with the inflammatory infiltrate and becomes more pronounced with the aging of the lesion. 8
CRDD has a good prognosis and usually has a spontaneous remission without treatment, but due to esthetic reasons or in relapsing cases, a great number of medical treatments can be applied with good results, with corticoid therapy being the most frequently used. In the case of isolated or small lesions, surgical excision is usually preferable. Other treatments used include radiotherapy, chemotherapy, and, more recently, some immunomodulators such as imatinib and methotrexate in small doses.5,8
Case Report
A white 57-year-old woman presented with a painless suppurative lesion on the right side of the neck, without any other associated symptoms. The patient did not present with a medical history of interest. During examination, a 1-cm erythematous nodular lesion on a base of slightly indurated subcutaneous tissue of approximately 3 cm was discovered (Figure 1). The patient did not present with adenopathies or visceromegalies. Therefore, an incisional biopsy was performed due to a suspicion of cutaneous adnexal tumor or lymphoma. The histopathologic study of the lesion showed a slightly acanthotic epidermis and a dense inflammatory infiltrate occupying the whole dermis and part of the subcutaneous cellular tissue made up of large histiocytes with abundant cytoplasm and vesicular nucleus, together with numerous lymphocytes, which sometimes formed aggregates, some multinucleated giant cells, plasma cells, and neutrophilic and eosinophilic polymorphonuclear cells. There were fibrocollagen bundles immersed in the infiltrate, which gave a certain fibrous aspect to the lesion. The characteristic phenomenon of emperipolesis (undamaged inflammatory cells in the cytoplasm of histiocytes) was also present. A histochemical study was performed for the screening of microorganisms (Silver, Ziehl Neelsen, periodic acid–Schiff [PAS], PAS-diastase, Giemsa, and Fite-faraco), which was negative. The immunohistochemical study showed S100-, CD68-, CD14-, and CD163-positive histiocytes; was focally positive for lysozyme; and was negative for AE1/AE3, CD1a, langerin, and factor XIIIa (Figure 2). These findings were compatible with CRDD, and after an extension study within normal limits (hemogram, biochemistry, erythrocyte sedimentation rate, kidney and liver function tests, proteinogram, immunoglobulins, complement, antinuclear antibodies, syphilis serology, hepatitis and human immunodeficiency virus, β2-microglobulin, and chest x-ray), the lesion was excised completely without relapse so far.
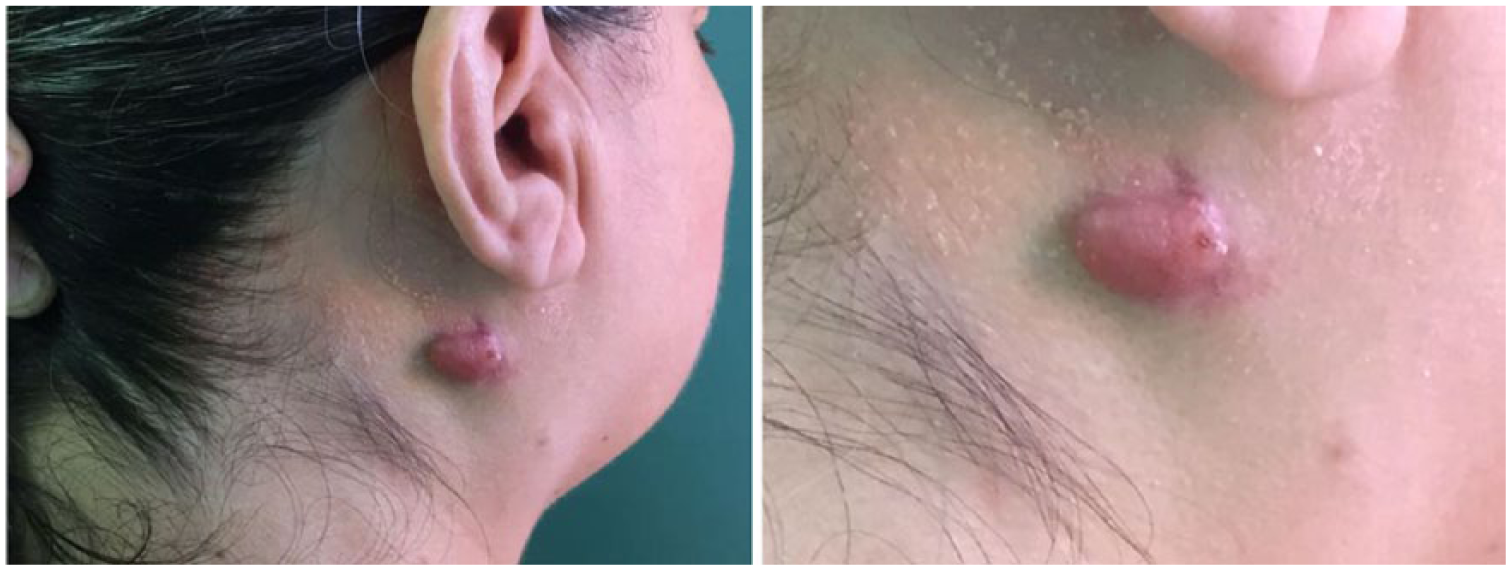
Erythematous nodule on an indurated base in the lateral cervical region.
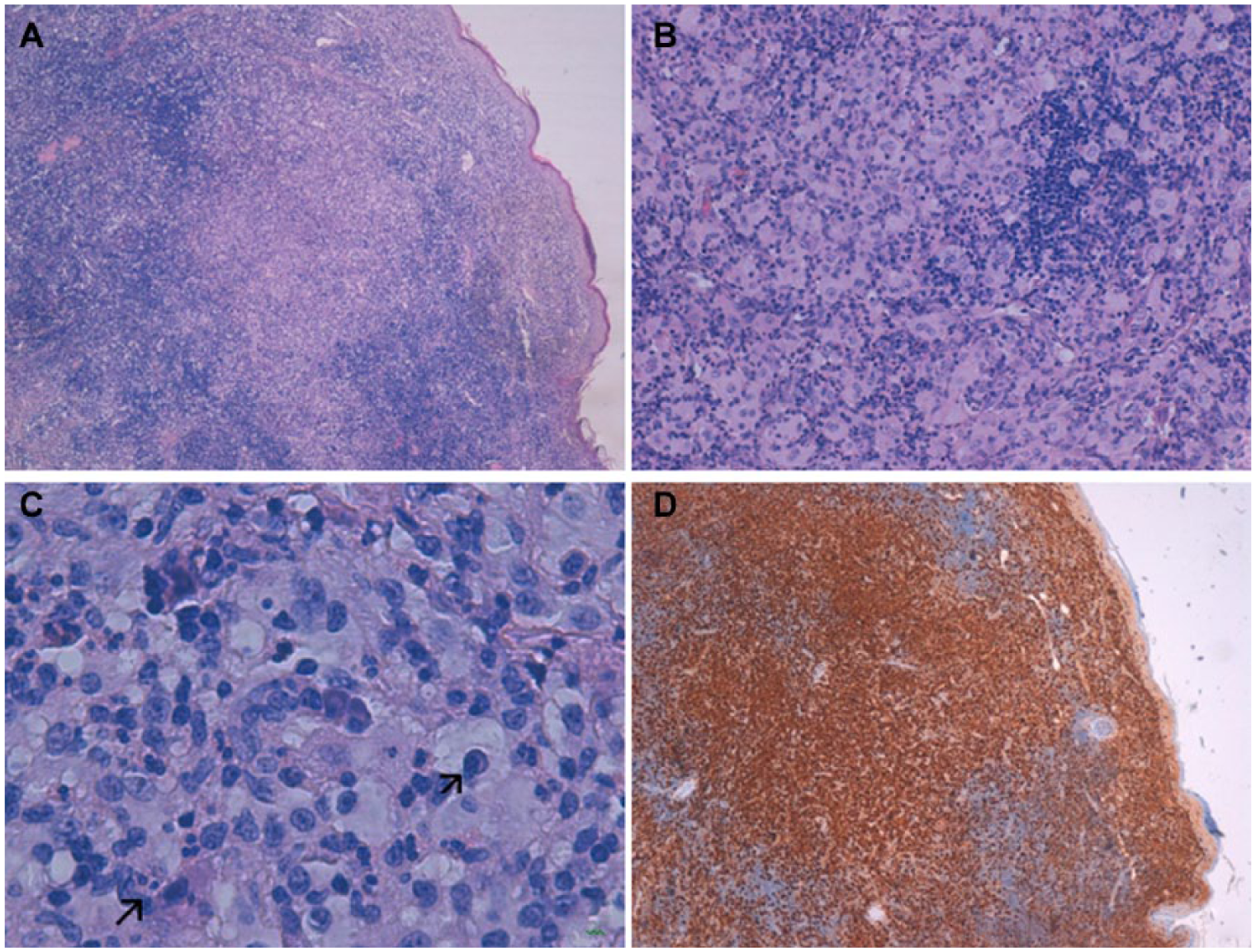
(A) Dense lymphohistiocytic infiltrate in the dermis (hematoxylin and eosin stain, ×40). (B) Large histiocytes with ample cytoplasm with soft edges and vesicular nucleus. Lymphoid aggregate on the right of the micrography (hematoxylin and eosin stain, ×200). (C) Plasma cell and neutrophil in the cytoplasm of a large histiocyte (emperipolesis) (hematoxylin and eosin stain, ×400). (D) Intense diffuse positivity of the histiocytic component with S100 immunohistochemical stain, with negative image of the lymphoid aggregates and the rest of inflammatory cellularity.
Discussion
CRDD is a rare clinical condition that can manifest in very different clinical ways and for whose diagnosis the histopathologic study of the lesions is crucial. Histologically, it is convenient to consider differential diagnosis with other histiocytic conditions such as Langerhans cell histiocytosis, in which histiocytes are smaller, the nucleus is kidney shaped or indented, there is epidermotropism, and there is immunohistochemical positivity for CD1a and langerin; reticulohistiocytosis, where histiocytes have a ground-glass-looking cytoplasm and there is usually immunohistochemical negativity for S100; juvenile xanthogranuloma, with vacuolated, xanthomatous, and fusiform histiocytes and a great number of Touton-type and foreign body–type multinucleated giant cells, which are negative or immunohistochemically focally positive for S100 and, unlike in CRDD, there is immunohistochemical positivity for factor XIIIa; and eruptive xanthoma, made up of aggregates of foamy-looking cells also with immunohistochemical negativity for S100. Besides, there is no emperipolesis in any of these conditions, although we should bear in mind that this phenomenon is not pathognomonic and can appear in some cases of B-cell lymphomas and in the cutaneous involvement of H syndrome. 8 Other disorders that should be ruled out are storage disorders, granulomatous diseases, histiocytic sarcoma, infections such as mycobacteriosis, histoplasmosis, and Kaposi sarcoma. 8 On the other hand, once the diagnosis of CRDD has been confirmed, it is important to perform a whole physical examination and an extension study to rule out systemic RDD.
CRRD usually has a favorable prognosis and can have spontaneous self-remittance; however, a treatment is usually applied with a good response to topical corticoids or a simple excision in the case of isolated or small lesions.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.