
Abstract
Cutaneous chronic graft-versus-host disease (cGVHD) is a frequent and debilitating complication of allogeneic hematopoietic cell transplantation. Skin involvement affects most patients and may involve extensive body surface area in severe cases. Manifestations range from inflammatory lichenoid eruptions to fibrotic and sclerotic changes that cause pain, functional impairment, and reduced quality of life. This review summarizes current understandings of the immunopathogenesis, clinical presentation, diagnostic tools, and management of cutaneous cGVHD. Systemic corticosteroids remain first-line therapy, but responses are variable and prolonged use causes substantial toxicity. Recently, therapeutic agents such as ibrutinib, ruxolitinib, belumosudil, and axatilimab, which modulate distinct immune and fibrotic pathways, improved management of this condition. Nevertheless, relapse and treatment-refractory disease remain common. Emerging strategies target immune dysregulation and fibrosis. Advances in scoring systems, imaging, and patient-reported outcomes are improving disease assessment. Given its heterogeneity, individualized, mechanism-based treatments are needed to improve long-term outcomes.
Keywords
Introduction
Cutaneous chronic graft-versus-host disease (cGVHD) remains a formidable challenge for patients and clinicians in the years following allogeneic hematopoietic cell transplantation (HCT). Despite advances in transplant protocols and supportive care, cutaneous involvement, the most common manifestation of cGVHD, continues to cause substantial morbidity and mortality, affecting 60% to 80% of patients.1-5 Growing evidence highlights the impact of skin disease on quality of life and long-term outcomes, underscoring the need to better understand cGVHD pathogenesis and develop novel therapies.
From a pathophysiologic standpoint, cutaneous cGVHD encompasses a wide spectrum of manifestations, ranging from erythematous, lichen planus-like/lichenoid rashes to sclerotic forms (sclerodermoid or eosinofilic fasciitis-like) that can severely compromise mobility. These manifestations arise from complex immune interactions among donor T cells, cytokines, and host antigens, driving persistent tissue damage. 6 Variable onset (insidious or abrupt) further complicates early diagnosis and standardized care. 7 Clinically, cutaneous cGVHD is broadly categorized into non-sclerotic (lichenoid, eczematoid, keratosis pilaris like, poikiloderma, leukoderma, etc.) and sclerotic phenotypes. Non-sclerotic disease includes lichenoid and eczematous variants, whereas sclerotic manifestations encompass lichen sclerosus-like, morphea-like, deep dermal sclerosis, and fascial involvement.
Prospective studies demonstrate that skin disease severity correlates with survival. 8 Of note, one 2023 multicenter cohort analysis documented that patients with active cGVHD had an increased risk of treatment-resistant disease and non-relapse mortality, highlighting the need for close dermatologic assessment and proactive intervention. Emerging data suggest improved diagnostic tools may enable earlier intervention. 4
Despite advances, treatment remains complex. Conventional therapy for cutaneous cGVHD relies on extended immunosuppression with corticosteroids and calcineurin inhibitors (CNIs), often associated with suboptimal response, adverse effects, or infections. 9 However, novel agents targeting B cells, T follicular helper cells, and specific cytokine pathways (eg, immunomodulation) are showing promise in ongoing clinical trials. 4 Furthermore, a growing emphasis on multidisciplinary care including dermatologic, rehabilitation, and supportive services holds potential for mitigating disease burden and improving patient-reported quality of life.
This review provides a clinically focused overview of adult cutaneous cGVHD, integrating current understanding of immunopathogenesis, phenotypes, diagnosis, and evolving therapies. We critically evaluate available evidence for both established and emerging treatments, highlight limitations in current management approaches, and identify areas requiring further investigation. Given its heterogeneity and progression, standardized assessment is essential for guiding therapy; the 2014 National Institutes of Health (NIH) Consensus Criteria (which is now >10 years old) remain the reference standard for grading disease severity and evaluating treatment response.10,11
Pathophysiology
Tissue Injury and Early Inflammation
In early cGVHD, tissue injury and inflammation initiate immune dysregulation and fibrosis (Figure 1). The preparative regimens that include high-dose chemotherapy and irradiation, cause direct cellular injury and disrupt epithelial barriers, leading to the release of damage-associated molecular patterns such as extracellular adenosine triphosphate (ATP). Extracellular ATP acts as a danger signal by binding P2X7 receptors on antigen-presenting cells (APCs), promoting costimulatory molecules (eg, CD80 and CD86) expression and interferon-γ secretion that drives donor T-cell expansion.5,12 Concurrently, the disruption of the mucosal barrier facilitates microbial translocation, thereby exposing pattern recognition receptors to pathogen-associated molecular patterns and further intensifying the inflammatory cascade. Transcriptomic analyses of cutaneous cGVHD lesions confirm activation of interferon-driven pathways, including STAT1- and TBX21-associated Th1 signaling, as well as upregulation of pattern-recognition receptor and phagocytic pathways, supporting the central role of innate immune activation in early disease.13,14 This environment activates endothelium and recruits donor immune cells into target tissues. Moreover, neutrophil recruitment, stimulated by chemokine gradients and microbial factors, amplifies tissue damage through the production of reactive oxygen species and proteolytic enzymes, which further deteriorate tissue integrity. 15 In parallel, early monocyte reprogramming has been demonstrated in preclinical models, where IL-17-dependent cGVHD is associated with transcriptional activation of circulating monocyte subsets enriched for NF-κB and Mitogen-Activated Protein Kinase (MAPK) signaling, indicating that systemic innate immune dysregulation precedes overt fibrosis. 16 In addition, early inflammatory signals contribute to the loss of regulatory T cell function, compromising peripheral tolerance mechanisms and perpetuating the cycle of tissue injury. 17 Histopathologic findings in affected tissues demonstrate vascular injury and immune cell infiltration in early disease, features that are thought to contribute to immune dysregulation and, over time, to the fibroproliferative changes characteristic of advanced cGVHD. 18
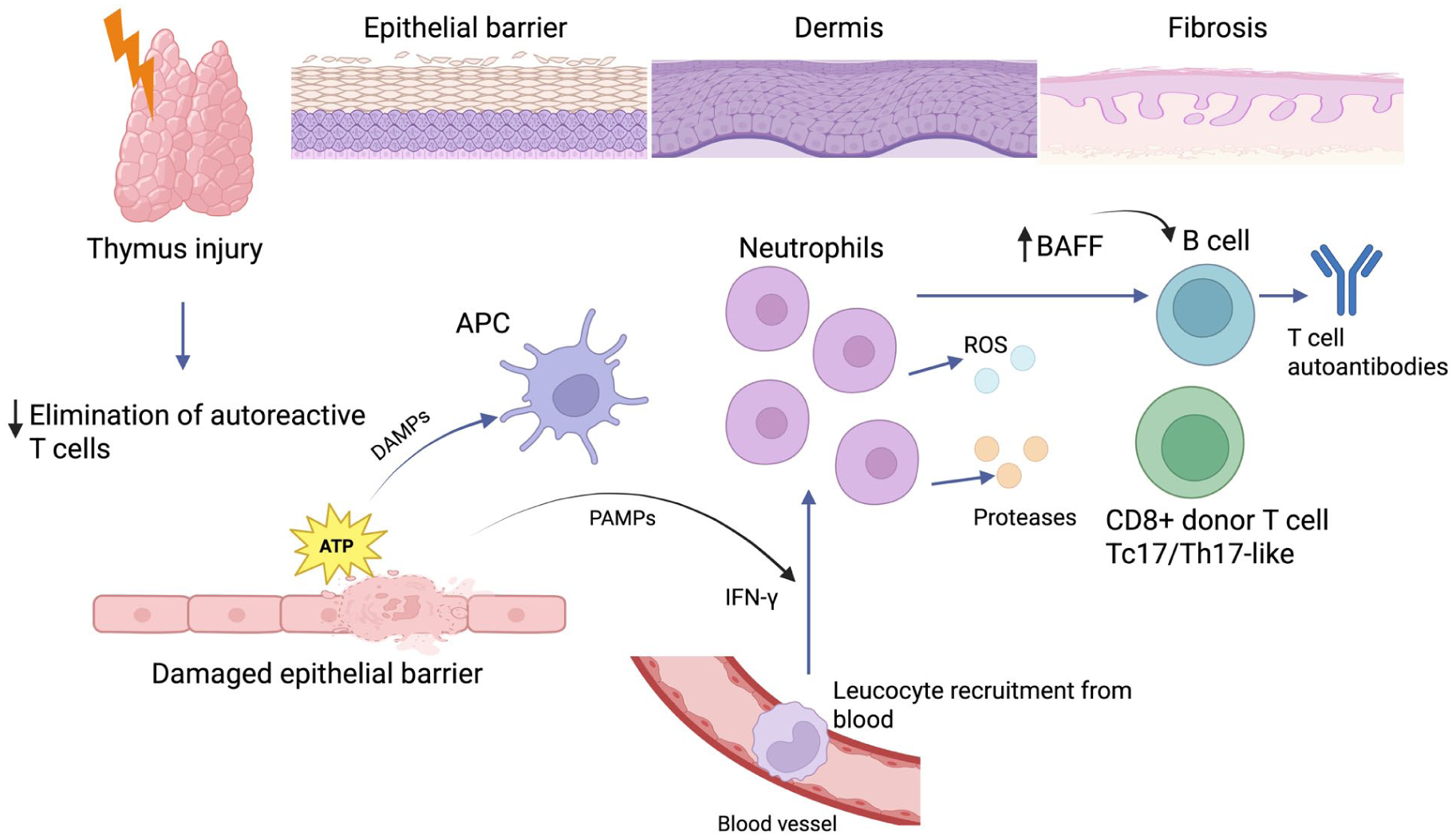
Pathophysiology of chronic cGVHD. The schematic illustrates the multi-step immunopathogenic cascade leading to tissue injury and fibrosis in chronic cutaneous GVHD. Damage to the thymus impairs negative selection of autoreactive donor-derived T cells, resulting in their persistence within the peripheral immune repertoire. Concurrently, epithelial barrier injury in the skin leads to the release of DAMPs such as extracellular ATP and the translocation of PAMPs from the microbiota. DAMPs engage purinergic receptors (notably P2X7) on APCs, while PAMPs further stimulate APC activation, promoting secretion of IFN-γ and other pro-inflammatory cytokines. Activated APCs and the cytokine milieu drive endothelial cell activation, enhancing adhesion molecule expression and vascular permeability, thereby promoting neutrophil recruitment into the dermis. Recruited neutrophils release ROS and proteolytic enzymes, amplifying local tissue damage and sustaining DAMP release. In parallel, donor-derived CD8+ T cells acquire a Tc17/Th17-like phenotype, producing IL-17 and other inflammatory mediators that perpetuate inflammation and tissue injury. B-cell activation, stimulated by elevated BAFF and supported by T-cell signaling, results in autoantibody production and further T–B cell cross-talk, which together promote fibroblast activation. These combined immune events culminate in fibroblast and myofibroblast proliferation, excessive extracellular matrix deposition, and cutaneous fibrosis, a hallmark of the chronic phase of the disease. APC, antigen-presenting cell; ATP, adenosine triphosphate; BAFF, B-cell activating factor; DAMP, damage-associated molecular pattern; IFN-γ, interferon-gamma; PAMP, pathogen-associated molecular pattern; ROS, reactive oxygen species; cGVHD, chronic graft-versus-host disease.
Dysregulated B-Cell and T-Cell Immunity
Thymic injury from conditioning and acute GVHD impairs central tolerance, allowing expansion of autoreactive T cells, reducing the expression of tissue-restricted antigens such as Class II-associated Invariant Chain Peptide (CLIP) that are crucial for eliminating autoreactive T cells during negative selection (Figure 1).19,20 This disruption permits generation of autoreactive T cells, while donor CD8+ T cells further damage medullary thymic epithelial cells and impair selection.21,22 In parallel, there is an expansion of a T helper 17 (Th17)-prone subset characterized by elevated RORγt expression and a mixed Th1/Th17 cytokine profile, including increased production of IL-17 and IFN-γ, which intensifies local inflammation and recruits additional effector cells to target tissues.23,24 Ribonucleic acid (RNA) sequencing of cutaneous cGVHD has demonstrated that lichenoid disease displays a mixed Th1/Th17 signature, whereas sclerotic disease is predominantly Th1-driven with a unique TSLP-OX40 axis. Strong enrichment of IFN-related gene expression is a shared feature of both cGVHD subtypes, while the TREM1 pathway appears specific to lichenoid disease.13,14 Moreover, IFN-inducible chemokines such as CXCL9 and CXCL10 are highly expressed in affected skin, supporting recruitment of CXCR3+ effector T cells and amplification of local inflammation. 13 Reduced thymopoiesis, as evidenced by lower T cell receptor excision circle levels, further impairs the generation of naïve T cells, exacerbating the dominance of pathogenic, autoreactive clones. Deficiencies in regulatory T cell populations, including conventional regulatory T cells, fail to restrain these aberrant responses, thereby perpetuating the cycle of autoimmunity and tissue damage. 25
B-cell dysregulation further fuels chronic inflammation. Activated B cells, acting as potent APCs, enhance the activation of T follicular helper cells and support the formation of ectopic germinal centers, where aberrant B-cell clonal expansion occurs. 26 This process leads to the production of pathogenic autoantibodies that contribute to tissue injury and fibrosis. Transcriptomic data from sclerotic cGVHD skin reveal upregulation of Fc receptor-associated and phagocytic pathways, suggesting that immune complex-mediated activation of macrophages may further amplify tissue inflammation. 13 In addition, elevated levels of B-cell-activating factors, such as B-cell activating factor (BAFF), provide survival signals that promote the persistence of autoreactive B cells, thereby reinforcing the chronic inflammatory state. 26 Failure to regulate B-cell responses, combined with deficient regulatory lymphocytes, sustains immune activation, autoantibody production, and end-organ damage central to cGVHD pathogenesis. 25
Fibrosis
Fibrosis in cGVHD results from a persistent, dysregulated wound-healing response in which prolonged immune activation drives extracellular matrix deposition and irreversible tissue remodeling. Initial inflammatory insults, together with sustained release of profibrotic cytokines such as transforming growth factor-β (TGF-β), create a microenvironment that favors fibroblast activation and differentiation into myofibroblasts. 27 Bulk RNA sequencing of sclerotic cGVHD skin demonstrates upregulation of canonical fibrotic pathways, including TGF-β-associated genes (TGFB1, TGFB3), collagen genes (COL3A1, COL11A1), fibronectin (FN1), and wound-healing signatures, confirming active matrix remodeling in affected tissues. 13 Moreover, distinct transcriptomic subsets have been identified within sclerotic cGVHD, including a fibroinflammatory subtype characterized by concurrent Th1 and fibrotic signaling and a predominantly fibrotic/TGF-β-driven subtype, suggesting biological heterogeneity in fibrogenic mechanisms. 13 Donor-derived macrophages, particularly those activated in a CSF-1-dependent manner, contribute to the establishment of a TGF-β-rich milieu that intensifies collagen deposition and sclerotic changes in target organs like the skin and lungs. 28 Preclinical single-cell analyses have shown that CSF-1 receptor-dependent monocyte/macrophage populations expand during IL-17-driven cGVHD and display transcriptional programs enriched for activation, chemotaxis, and inflammatory signaling, linking systemic monocyte dysregulation to tissue fibrosis. 16 In addition, IL-17-dependent transcriptional signatures in circulating monocytes can be detected before clinical diagnosis of cGVHD becomes apparent, supporting a role for early systemic immune reprogramming in subsequent fibrotic evolution. 16
Concurrently, dysregulated B-cell immunity plays a pivotal role in sustaining fibrosis through disrupted B-cell homeostasis and autoantibody production. Excess levels of BAFF rescue autoreactive B cells from peripheral deletion, enabling these cells to persist and become activated, which in turn leads to the production of pathogenic antibodies that form immune complexes and stimulate the release of additional fibrogenic cytokines. 29 Altered B-cell subsets, marked by a reduction in naïve B cells and a proportional increase in activated or plasmablast-like cells, further contribute to chronic inflammatory signals that drive collagen synthesis.30,31 This breakdown in normal B-cell tolerance, compounded by the impact of autoantibody formation, reinforces a self-sustaining cycle of inflammation and fibrosis that ultimately can culminate in the scleroderma-like features observed in cGVHD. 25
The skin is the largest and most immunologically active barrier organ, containing abundant resident dendritic cells, macrophages, and mast cells, that facilitate alloimmune recognition and effector cell recruitment. Transcriptomic analyses confirm enrichment of Th1 signaling, macrophage activation pathways, neuroinflammatory signatures, and TGF-β-driven fibrotic programs within sclerotic skin lesion.13,14 Donor-derived T cells and macrophages, particularly those activated in a CSF-1-dependent manner, infiltrate the skin and drive local production of profibrotic cytokines such as TGF-β and IL-17, leading to fibroblast activation and extracellular matrix deposition.28,32-35 Keratinocytes, the main targets of donor T cells, can amplify fibrosis by producing TGF-β1 in response to IFN-γ and apoptotic signals, further linking inflammatory and fibrotic pathways. 35 Mast cells also contribute to fibrosis and effector cell recruitment in the dermis. 33
Although photoexposure and other local insults can precipitate or localize cGVHD lesions, these are not the primary reason for the skin’s overall predominance as a target organ. Instead, the skin’s barrier function, immune cell composition, and regenerative dynamics make it especially vulnerable to the dysregulated immune responses and maladaptive repair that characterize cGVHD.5,25,36 Table 1 summarizes currently recognized risk factors for cGVHD highlighting their molecular significance.
Risk Factors for Cutaneous cGVHD with Proposed Mechanisms.
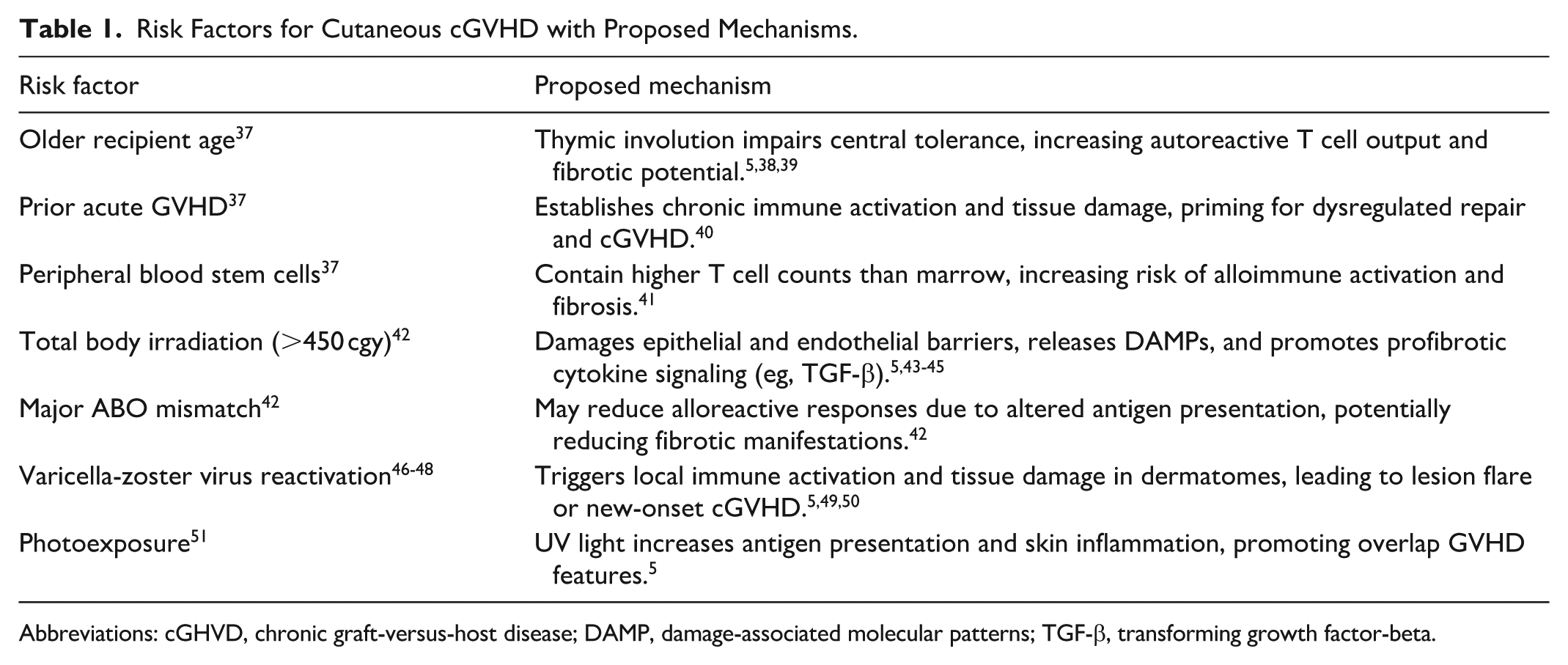
Abbreviations: cGHVD, chronic graft-versus-host disease; DAMP, damage-associated molecular patterns; TGF-β, transforming growth factor-beta.
Classification of Cutaneous Lesions
Cutaneous GVHD spans a broad clinical and histopathologic spectrum rooted in the complex interaction between donor T (and sometimes B) cells and host target tissues. Clinically, GVHD is differentiated into acute and chronic forms, determined less by rigid timing than by distinctive morphologies and immunologic underpinnings. The 2014 NIH Consensus emphasized classifying cGVHD into “conventional” (classic) cGVHD and “overlap” cGVHD, reflecting the varied patient presentations observed in practice. Classic cutaneous cGVHD includes both non-sclerotic (lichenoid, eczematoid, keratosis pilaris like, psoriasiform) and sclerotic phenotypes. 52
Cutaneous acute GVHD commonly appears as erythematous maculopapular eruptions that may evolve into confluent or bullous lesions resembling toxic epidermal necrolysis. 53 Palmar-plantar or other acral site (ears, nose, etc.) involvement, mucosal changes, or parallel gastrointestinal and hepatic dysfunction (for instance, diarrhea or hyperbilirubinemia) strengthens the clinical diagnosis of acute GVHD. 54 Histologically, prominent vacuolar interface changes in the basal layer, keratinocyte apoptosis, and interface dermatitis together with clinical presentation and timing of the eruption help confirm the diagnosis of acute GVHD in the right clinical setting. 27
The clinical differential diagnosis of acute graft-versus-host disease (aGVHD) most commonly includes drug eruption, viral exanthem, toxic erythema of chemotherapy, engraftment syndrome and cutaneous eruption of lymphocyte recovery. All conditions usually demonstrate mild vacuolar interface dermatitis on histopathology, making distinction based solely on microscopic findings difficult. The presence of abundant eosinophils favors a drug eruption; however, a few eosinophils do not exclude aGVHD, whereas their complete absence argues against a drug-induced process. “Satellite cell necrosis,” a classic but non-specific clue for GVHD, may also occur in herpetic dermatitis and other inflammatory conditions. Biopsy confirms interface dermatitis compatible with aGVHD, requiring clinical correlation for diagnosis.55-58
In chronic GVHD, histopathologic findings are variable and classically fall into lichenoid, sclerotic, or, less commonly, eczematous patterns. The lichenoid type resembles idiopathic lichen planus, exhibiting band-like lymphocytic inflammation that obscures the dermo-epidermal junction, irregular epidermal hyperplasia with “saw-tooth” rete ridges, and wedge-shaped hypergranulosis. The sclerodermoid variant shows dense dermal collagen with an atrophic overlying epidermis and little or no remaining interface change, including basal layer apoptosis and vacuolar change, as recent analyses have shown that interface activity is common even in sclerotic cGVHD. 59 A third, eczematous pattern is characterized by spongiotic dermatitis and chronic inflammatory infiltrates. Because these features overlap with those of lichen planus, morphea/scleroderma, and eczema, clinicopathologic correlation remains indispensable for diagnostic accuracy.55,58
Conventional cutaneous cGVHD can arise even before day 100 post-transplantation if “diagnostic” associated lesions, such as lichen planus-like papules, lichen sclerosis & atrophicus or morphea-like sclerotic plaques, are noted.10,52 Lichenoid cGVHD frequently presents with violaceous papules and plaques accompanied by pruritus and potential scaling, whereas sclerotic cGVHD can lead to extensive skin thickening and restricted joint mobility, especially, when the fat or fascial layer is involved.53,60 Histology ranges from focal interface changes with apoptotic keratinocytes, seen in both lichenoid and many sclerotic lesions, to markedly thickened collagen in advanced sclerotic disease. 59 Overlap cGVHD displays both chronic and acute-type patterns, portending worse outcomes and heightened functional impairment that generally necessitates more aggressive immunosuppressive therapy.10,61,62
Distinguishing acute, chronic, and overlap cGVHD guides therapy. Sclerotic cGVHD, in particular, may demand both pharmacologic and rehabilitative interventions to preserve range of motion and reduce tissue fibrosis.53,60
Clinical Presentation
Cutaneous cGVHD is clinically heterogeneous and often mimics autoimmune and autoinflammatory skin diseases. It remains unclear whether this diversity reflects discrete aberrations across multiple immune pathways or the protean manifestations of a single underlying dysregulation. 8 According to the NIH consensus criteria (Table 2), the spectrum of cutaneous cGVHD can be broadly classified into nonsclerotic (ie, predominantly papulosquamous) and sclerotic subtypes. Nonsclerotic disease, characterized by inflammatory eruptions such as morbilliform, lichen planus-like, or papulosquamous rashes without fibrosis, typically presents earlier after transplant, with 1 study reporting a median onset of ~9 months and is generally more responsive to treatment when assessed by the extent of body surface area (BSA) involvement.10,63
Consensus Criteria for cGVHD: Dermatologic Involvement.

In contrast, sclerotic cutaneous cGVHD, which encompasses both superficial and deep fibrotic changes, tends to occur later, with a median onset at ~19 months, and is associated with progressive, irreversible tissue damage/remodeling that contributes significantly to morbidity. 63 Notably, recent findings indicate that in patients with skin of color (SOC), the cosmetic and psychosocial impacts of dyspigmentation associated with nonsclerotic disease may impose an even greater quality of life burden than the morbidity traditionally attributed to sclerotic changes. 64 These findings underscore the interplay between inflammation and fibrosis and the need for individualized management.8,10,63,64
Nonsclerotic Manifestations
Nonsclerotic cutaneous cGVHD is a highly heterogeneous entity that encompasses a broad range of clinical morphologies, which is why the clinical community now prefers the term “nonsclerotic” over the prior/legacy nomenclature of “lichenoid GVHD.” 5 One of the most common presentations is the lichen planus-like variant, in which patients develop well-demarcated, pink to purple/violaceous papules and plaques with lacy skin changes that closely resemble idiopathic lichen planus, yet tend to be more extensive and involve lips/oral mucosa and atypical sites including face, neck, palms, and soles. 64 In individuals with darker skin tones/SOC, the conventional erythema is often masked by baseline pigmentation and may instead present as a grayish hue, making subtle inflammatory changes more challenging to discern. 11
Beyond lichen planus-like lesions, nonsclerotic cGVHD may also manifest as poikiloderma, psoriasis-like eruptions, eczema-like lesions, or keratosis pilaris-like changes, all of which frequently begin with symptoms of dry, scaly, and intensely pruritic skin.66,67 While primary care physicians may consider using antihistamines for the management of itch, their use should be avoided in cGVHD patients as they can exacerbate existing sicca symptoms in this population. As the acute inflammatory phase subsides, residual dyspigmentation and ichthyosis often emerge as post inflammatory changes; although these features reflect prior damage rather than active disease, they can be cosmetically disfiguring and significantly impact quality of life. 68
Adnexal involvement is common, with hair changes ranging from nonscarring alopecia—characterized by scalp scaling, pruritus, and diffuse thinning—to scarring (cicatricial) variants, including lichen planopilaris-like alopecia that may result in permanent follicular loss.69,70 Nail abnormalities range from mild longitudinal ridging and splitting to severe dystrophy/pterygium that may interfere with daily function (Table 2).69,70 Moreover, nonsclerotic cutaneous cGVHD can mimic various autoimmune conditions such as vitiligo/leukoderma, alopecia areata, cutaneous lupus erythematosus, and even dermatomyositis, blurring the lines between distinct cGVHD pathology and other autoimmune phenomena.71-73
Interestingly, additional studies have reported that adnexal involvement, including vitiligo and alopecia areata, may correlate with more severe disease course, suggesting that nonsclerotic manifestations may herald a broader dysregulation of the immune system post-transplant.74,75 These presentations illustrate the complexity of nonsclerotic cGVHD. 72
Sclerotic Manifestations
Sclerotic manifestations of cGVHD can involve both the superficial and deeper layers of the skin and may arise de novo or evolve as a progression from non-sclerotic lesions.8,25,59,65,76 Superficial sclerotic changes are typically characterized by lichen sclerosus-like plaques that typically present as well-demarcated, ivory, and shiny plaques/patches with cigarette paper like atrophy and/or follicular plugging sometimes appearing in a setting of poikiloderma (Table 2).59,76 These changes are readily appreciated on visual inspection and may also be detected on palpation, with reduced skin pliability or elasticity compared to adjacent uninvolved skin. 9 In contrast, deeper fibrosis presents as morphea-like or eosinophilic fasciitis (EF)-like lesions that may extend into the reticular dermis, subcutaneous tissue (eg, panniculitis), and fascia (eg, eosinophilic fasciitis).59,76,77 While in idiopathic forms, morphea and EF generally start as round or oval plaques that gradually become indurated and firm, in cGVHD plaques are often ill defined and more widespread.76,78 Early morphea-like and/or EF-like cGVHD is challenging to diagnose as early lesions present with cellulitis-like edema and/or violaceous erythema (depending on the depth of involvement).76,79 Once fibrosis sets, the skin becomes tight, difficult to pinch and often hyperpigmented in the center with or without lilac halo or peripheral erythema. When skin overlying joints is affected, the risk of joint motion restriction and functional limitation is high necessitating prompt and aggressive therapies. (Supplemental Appendix 1)
Histopathologically, sclerotic cGVHD is distinguished by a homogenized, edematous collagen band with a loss of elastic fibers and only minimal inflammatory infiltrate, features that distinguish it from other fibrosing disorders such as systemic sclerosis. 76 Clinical series have also described variants where sclerotic changes are associated with features resembling hypertrophic lupus erythematosus and eosinophilic fasciitis, further expanding the recognized spectrum of sclerotic GVHD. 80 Although sclerotic cGVHD more commonly affects the trunk and extremities, involvement of the facial and buccal fascia can occur and may result in reduced oral aperture and perioral tightness. In contrast, genital tract involvement is common yet often underrecognized, necessitating thorough examination and prompt referral to gynecology or urology.9,80,81 These sclerotic alterations lead to significant functional impairments, including joint contractures and chest wall rigidity that can result in pulmonary restriction, as well as chronic ulcers, angiomatosis, calcinosis, and an elevated risk for cutaneous squamous cell carcinoma (cSCC), which in this setting is best described as Marjolin Ulcer82,83 and has distinct pathgenesis and high-risk/poor prognosis, when compared to conventional ultraviolet-driven cSCC. Early recognition and management may mitigate fibrosis progression.1,8,76,80,84
Considerations for SOC Patients
In patients with richly pigmented skin, conventional findings such as erythema may be subtle or absent, and cGVHD may manifest as dyspigmentation (ash-color, hypo- or hyperpigmentation), poikiloderma, or lichen planus-like violaceous papules that may be less apparent than in lighter skin (Table 2). Sclerotic changes may present as areas of induration, loss of skin markings, or shiny, bound-down plaques, but the degree of visible sclerosis may be underestimated in the SOC.25,85,86
Histopathologic features of cutaneous cGVHD appear broadly similar across skin phototypes, with epidermal/interface changes (eg, basal keratinocyte apoptosis, vacuolar change, and lymphocytic satellitosis) commonly observed in epidermal cGVHD, and sclerosis affecting the dermis and sometimes subcutis/fascia in sclerotic disease.59,86 In a retrospective cohort of Korean patients, lichenoid patterns predominated clinically and histologically, with sclerodermoid changes and acute/chronic overlap was also reported.59,86 Data specifically evaluating histopathologic differences by SOC remain limited; however, pigment incontinence and dermal melanophages may be more conspicuous in lichenoid/interface patterns and may contribute to clinically apparent dyspigmentation.
Diagnosis
The diagnosis of cGVHD is based on the 2014 NIH consensus criteria, which integrate clinical findings, pathology, and additional testing to confirm the disease and assess its severity. 10 The criteria classify clinical features into 4 groups: (1) diagnostic, which independently establish cGVHD; (2) distinctive, which require further histologic or laboratory confirmation; (3) other, which are considered cGVHD-related only if another organ is involved; and (4) common, which are observed in both acute and chronic GVHD (Table 2). The diseases can be considered as being mostly superficial (affecting dermal epidermal junction), deep dermal or subcutaneous or consideration can be given whether disease is limited to skin or involves mucosa and/or adnexal structures, hair or nails. 10 This depth assessment is critical when considering skin directed therapies (eg, topical product strength or use of ultraviolet B vs ultraviolet A phototherapies). A multinational survey of 72 transplant centers reinforced the necessity of skin biopsy when cutaneous cGVHD is suspected but lacks clearly diagnostic features. 62
Given its multisystem impact, cGVHD should be managed in specialized transplant centers.75,87 This collaborative approach improves treatment decisions, enhances patient care, and helps prevent long-term complications. Experience from a dedicated GVHD-focused multidisciplinary clinic demonstrates that coordinated evaluation by transplant physicians, dermatologists, oral specialists, physiatrists, and nutritionists enables systematic organ assessment according to NIH criteria and structured range-of-motion evaluation, which is particularly important for detecting and monitoring sclerotic involvement. 88 Integration of patient-reported outcome (PRO) measures within this model has shown a high baseline symptom burden and a significant reduction in distress over follow-up, supporting the benefit of specialized, coordinated care. Additionally, incorporation of telehealth into a multidisciplinary framework expanded access to patients living farther from transplant centers and improved continuity of care without compromising comprehensive assessment. 88
A comprehensive physical examination is essential in diagnosing cutaneous cGVHD, requiring careful visual inspection and palpation of the skin to assess texture, thickness, color and any abnormal changes. Evaluation should include full skin and mucosal surfaces exposure to detect early or subtle manifestations of the disease. Special attention is given to areas prone to sclerosis, such as the extremities, trunk, genitalia, and joints, where restricted movement, deep subcutaneous fat or fascial involvement, and contractures may develop. Abduction and external rotation of the shoulders and hips can help identify grooving and cellulite-like changes, which suggest fascial involvement. Additionally, the GVHD Photographic Range of Motion assessment can help quantify joint stiffness and contractures, aiding in the early detection of functional impairment. 10 For joint range of motion, goniometer can prove indispensable for objective measurement.
When clinical signs alone are insufficient for a definitive diagnosis, skin biopsy is recommended, particularly in cases where distinguishing nonsclerotic cGVHD from other inflammatory dermatoses may be challenging. 89 A 4-mm punch biopsy is typically adequate for nonsclerotic presentations. For suspected sclerotic disease involving the dermis or subcutaneous fat, a 5 to 6 mm punch biopsy extending to the subcutis is preferred. 90 However, standard punch biopsy specimens frequently do not capture the fascia, limiting their utility in the evaluation of deep fascial involvement, as recently demonstrated in histopathologic analyses of cGVHD. 59 In cases where eosinophilic fasciitis-like disease is strongly suspected, deeper incisional or telescoping biopsy techniques may be required. 91
Recognizing mucosal disease can support the diagnosis of cutaneous cGVHD, particularly when distinctive features, such as lichenoid lesions in the oral mucosa, are present. 85 It is further critical to examine ocular, oral and genital mucosa for signs of cGVHD (Table 2)
Diagnosing sclerotic and fascial cGVHD presents a challenge due to its gradual progression and resemblance to other lower extremity disorders (eg, lipodermatosclerosis). 92 Magnetic resonance imaging (MRI) has emerged as a valuable noninvasive diagnostic tool for assessing deep tissue involvement, particularly in cases where the skin appears unaffected but functional impairment, restricted mobility, or fascial thickening is suspected. 92 MRI can reveal pathological changes in the dermis, subcutaneous fibrous septa, fascia, and muscle, with short tau inversion recovery sequences being especially useful for identifying soft-tissue edema indicative of active disease.
Beyond MRI, several emerging technologies have been explored for diagnosing and monitoring sclerotic cGVHD. High-frequency ultrasound can evaluate skin and subcutaneous tissue thickness, helping differentiate active inflammation from fibrosis. 93 Digital heat mapping, which measures temperature variations in affected areas, has been investigated for its potential to detect increased vascular activity associated with inflammation. 93 Additionally, myotonometry and durometry, which assess tissue stiffness and elasticity, have been studied as potential tools for quantifying fibrosis severity and disease progression. 93 These modalities may complement MRI by offering quantifiable, objective measures of disease activity without requiring invasive procedures. 93 While these techniques hold promise, none have been validated for routine clinical use, and their role in standard diagnostic and monitoring protocols remains under investigation. 94
Biomarker research continues to advance, with studies identifying BAFF, CXCL10, and CXCL11 as potential indicators of cGVHD activity. 94 However, a reliable, universally accepted biomarker for sclerotic cGVHD is yet to be established, reinforcing the need for multidisciplinary clinical assessment, imaging, and histopathological correlation for accurate diagnosis. 95 As research advances, combining advanced imaging, biomechanical assessment, and biomarker profiling may help refine early detection and personalized treatment strategies for sclerotic and fascial cGVHD. 95
Grading System for Cutaneous cGVHD
The grading of cutaneous cGVHD relies primarily on the NIH Consensus Criteria, which were first introduced in 2005 and subsequently updated in 2014. 96 This system remains the most widely used and standardized approach for assessing the severity of cGVHD across multiple organ systems. The NIH criteria employ a four-point grading scale (0-3), where 0 indicates no involvement, 1 represents mild disease, 2 indicates moderate severity, and 3 corresponds to severe disease with significant functional impairment. 97 For cutaneous cGVHD, key features incorporated into the grading system include BSA involvement, the presence of erythema or violaceous skin changes, sclerosis, panniculitis, fascial restriction, ulceration, and the extent of genital, oral, and ocular involvement. 98 Skin findings vary in SOC patients, where erythema is less prominent. These parameters enable clinicians to categorize global disease severity as mild, moderate, or severe, facilitating consistent clinical assessments and therapeutic decision-making. 99
Patients with mild disease typically exhibit limited skin involvement (<18% BSA, following “The Rule of Nines” assessment) without significant functional disability. Moderate disease involves more extensive skin manifestations (18%-50% BSA) and may affect multiple organs, though without critical impairment. Severe disease is characterized by widespread skin involvement (>50% BSA), deep sclerotic features, erosions/ulcers and/or functional limitations such as restricted joint mobility due to fascial sclerosis. 96
In addition to the NIH clinical grading criteria, adjunct PRO measures such as the Lee Symptom Scale (LSS) and the SF-36 have been explored to complement the assessment of cGVHD severity and better capture symptom burden and quality of life impact.100-102 The LSS, a validated PRO measure, is frequently used to quantify the symptomatic burden of cGVHD across different organ systems (Supplemental Appendix 2). 103 Additionally, the Short Form-36 (SF-36), a general health-related quality of life instrument, has been widely applied in cGVHD research and clinical trials (Supplemental Appendix 3). 104 In some cases, the Modified Rodnan Skin Score (mRSS), originally developed for systemic sclerosis, has been used to assess skin thickening and fibrosis in cGVHD patients (Supplemental Appendix 1). 98 However, these methods have not outperformed the NIH scoring system in predicting clinical outcomes. 105
Recent advancements in technology-assisted and imaging-based methods have sought to improve the objectivity and reproducibility of cGVHD grading. Artificial/augmented intelligence (AI)-based algorithms have demonstrated promising accuracy in analyzing skin photographs to quantify erythema and sclerosis, offering an automated and standardized approach to disease assessment. 106 Additionally, 3D photography and hyperspectral imaging have been introduced to enhance BSA mapping and segmentation of affected skin, allowing for more precise measurements.107,108 These imaging technologies have been particularly useful in distinguishing active versus “burned out” sclerotic cGVHD. 109 Some studies have also explored the utility of skin punch biopsies to measure changes in fibrosis depth over time, though these remain an adjunct rather than a primary grading tool. 99
Furthermore, automated computer-aided image segmentation has shown promise in improving the detection of erythema and sclerosis in cGVHD patients. AI models trained on patient images have demonstrated the ability to identify subtle disease activity that may be overlooked during routine clinical evaluations. 105 One study suggested that erythema scoring is a potential prognostic marker, with higher erythema levels correlating with poorer survival outcomes in cGVHD. 106 Unfortunately, though, erythema can be difficult to assess in SOC patients. While these technological innovations are promising, further validation in large-scale clinical studies is required before they can be fully integrated into routine practice. 107
Despite these advances, challenges remain in achieving widespread standardization of non-invasive imaging techniques and AI-driven diagnostic tools. While the NIH criteria continue to serve as the cornerstone of cGVHD assessment, incorporating novel PRO measures, AI-powered image analysis, and advanced imaging modalities may enhance diagnostic accuracy, enable early detection of disease progression, and facilitate more precise therapeutic interventions. 109
Management
The management of cutaneous cGVHD can be broadly divided into topical/skin-directed and systemic treatments (Tables 3 and 4). The choice of treatment is influenced by disease severity, extent and depth of skin involvement, and whether other organ systems are affected. Topical treatments, such as corticosteroids (TCS) and calcineurin inhibitors (TCI), are traditionally the first-line approach and can be initiated promptly. In cases where the disease is more widespread or refractory, field skin-directed therapies (eg, phototherapy) and systemic therapies may be necessary, requiring careful consideration of infection risk, symptom burden, ease of access (eg, extracorporeal photopheresis (ECP)), quality of life impact, and potential for permanent tissue damage.
Comparative Efficacy of Systemic Therapies and ECP in cGVHD.
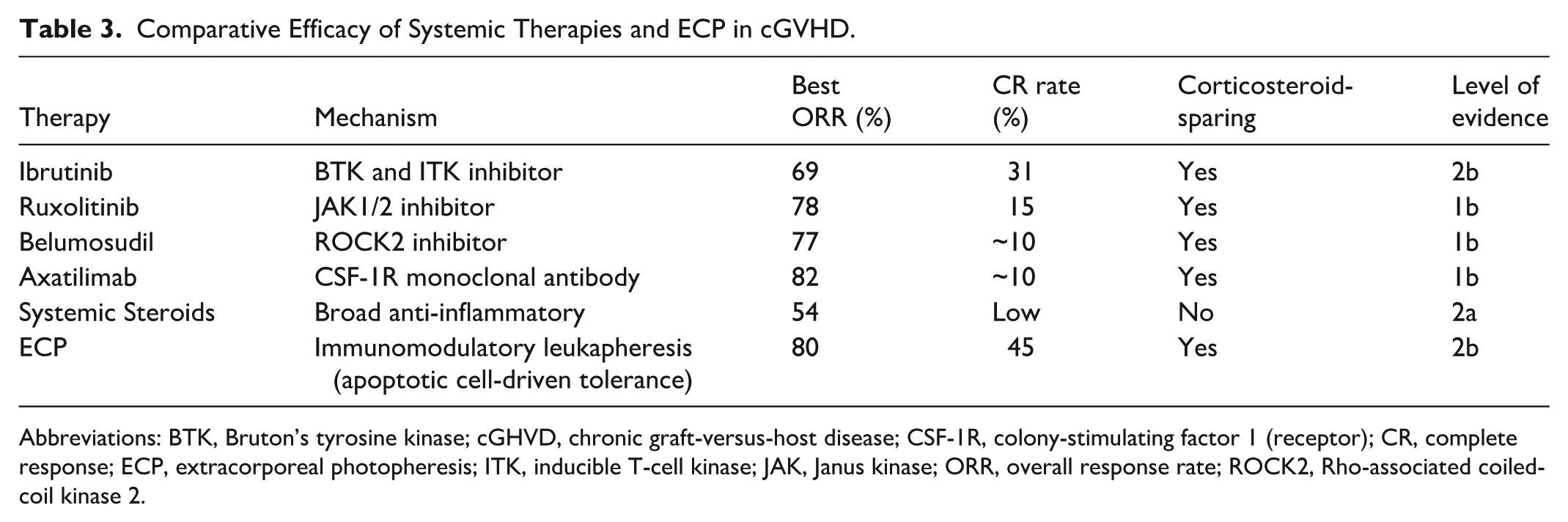
Abbreviations: BTK, Bruton’s tyrosine kinase; cGHVD, chronic graft-versus-host disease; CSF-1R, colony-stimulating factor 1 (receptor); CR, complete response; ECP, extracorporeal photopheresis; ITK, inducible T-cell kinase; JAK, Janus kinase; ORR, overall response rate; ROCK2, Rho-associated coiled-coil kinase 2.
Systemic Therapy Recommendations for Cutaneous cGVHD.
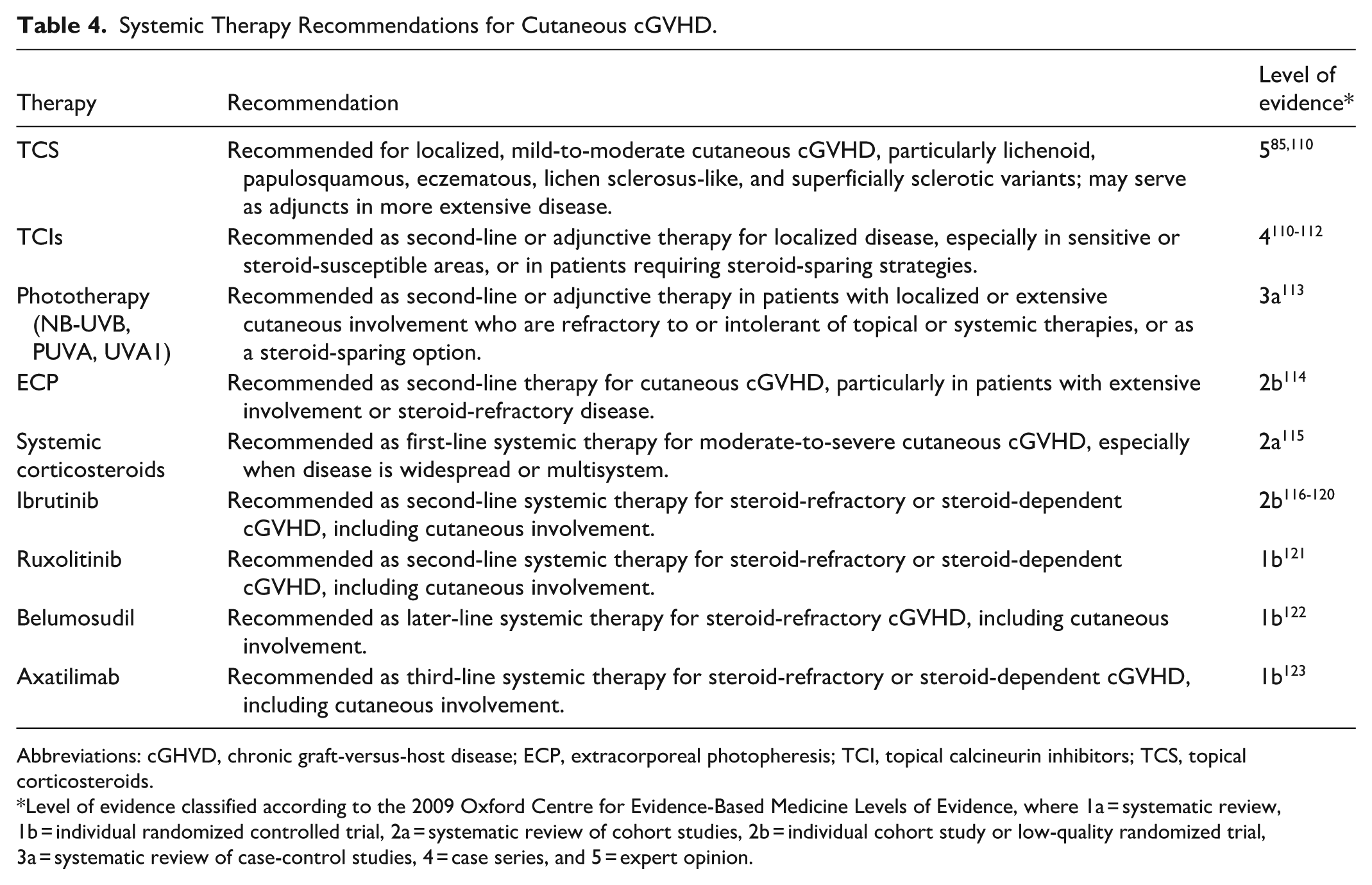
Abbreviations: cGHVD, chronic graft-versus-host disease; ECP, extracorporeal photopheresis; TCI, topical calcineurin inhibitors; TCS, topical corticosteroids.
Level of evidence classified according to the 2009 Oxford Centre for Evidence-Based Medicine Levels of Evidence, where 1a = systematic review, 1b = individual randomized controlled trial, 2a = systematic review of cohort studies, 2b = individual cohort study or low-quality randomized trial, 3a = systematic review of case-control studies, 4 = case series, and 5 = expert opinion.
General supportive measures for patients with cutaneous cGVHD are essential adjuncts to pharmacologic and procedural therapies. Regular skin checks to monitor for skin cancer/Marjolin Ulcer (vs angiomatosis) are critical, as patients with cGVHD—especially those receiving long-term immunosuppression or photochemotherapy—are at increased risk for both melanoma and non-melanoma skin cancers. Dermatologic surveillance should be performed at least annually, and more frequently in high-risk individuals.25,85,124
Sun avoidance is strongly recommended, as ultraviolet exposure can exacerbate cGVHD and further increase skin cancer risk. Patients should use broad-spectrum sunscreens (sun protection factor ≥ 30), wear protective clothing, and avoid peak sunlight hours. The use of moisturizers is a cornerstone of supportive care. Liberal, regular application of bland emollients helps restore barrier function, reduce pruritus, and prevent fissuring and secondary infection. This is particularly important in sclerotic or xerotic skin, where barrier compromise is common.85,124,125 As highlighted earlier, antihistamines are best avoided in cGVHD patients due to coexisting sicca symptoms.
Strategies to avoid friction and trauma are crucial in sclerotic cGVHD, as scarred or sclerotic skin has significantly reduced tensile strength and is prone to ulceration/erosions and secondary infection. Patients should avoid tight clothing, repetitive trauma, and activities that may cause shearing or pressure injuries even as simple as carrying a heavy bag on a shoulder strap.85,124
Skin-Directed Therapies
Topical Therapies
Corticosteroids
TCS serve as the first-line therapy for mild or localized cases of cutaneous cGVHD. They are particularly effective for managing limited papulosquamous, eczematous, lichen planus-like, lichen sclerosus-like, and superficial sclerotic lesions (<10%-15% BSA). In cases of moderate to severe disease (>15% BSA), they are often used in combination with field or systemic therapies to enhance symptom relief and control inflammation. 111
TCS (Supplemental Table 1) are available in varying strengths and formulations, and the choice of agent is guided by disease severity and the anatomical site of involvement. For thicker or more resistant lesions or involvement of hands/palms, soles, scalp, higher-potency corticosteroids may be required, while lower-potency formulations are preferred for delicate areas such as the face, genitalia, and intertriginous zones. 111 Medium-potency formulations may be appropriate for involvement of the trunk and extremities. Treatment is typically administered in limited courses, with intermittent application strategies often used.
While TCS provide effective symptom control, prolonged use can lead to adverse effects, including cutaneous atrophy, striae, telangiectasias, and secondary infections. To mitigate these risks, it is recommended that patients avoid prolonged continuous application and use intermittent regimens when appropriate. If atrophic changes occur, transitioning to lower-potency steroids or alternative treatments is advised. 111
Calcineurin Inhibitors
TCIs, such as tacrolimus (0.1% ointment) and pimecrolimus (1% cream), are valuable alternatives to corticosteroids, particularly in sensitive areas where steroid-induced atrophy should be avoided. These agents are immunosuppressants that inhibit calcineurin, thereby preventing T cell activation and inflammatory cytokine release.111,112
TCIs have shown efficacy in reducing erythema/inflammation, pruritus, and scaling in cGVHD patients, and are used routinely in combination with TCS since their toxicity profiles do not overlap. 112 Studies have demonstrated that topical tacrolimus improves skin manifestations in approximately 72% of patients within hours to days, making it an effective adjunct to systemic therapy. 112 Additionally, its lack of effect on collagen synthesis means it does not contribute to skin atrophy, making it a safer option for prolonged use on facial and genital regions. 111 However, caution is required when using tacrolimus over large BSAs. Cases of systemic absorption leading to toxic tacrolimus levels have been reported, particularly when occlusive dressings or large application areas (>50% BSA) are involved. 126 Skin in cGVHD due to inflammation, xerosis and lichenification may lose its effective barrier function leading to greater absorption of TCIs. Serum monitoring may be necessary in cases where extensive topical tacrolimus use is required.
While TCS and TCIs are effective for superficial and inflammatory forms of cGVHD, they provide limited benefit in deeply sclerotic disease as they don’t penetrate into deeper-seated tissues. 112 Despite their widespread use, corticosteroids alone or in combination with other immunosuppressants frequently fail as first-line therapy for cGVHD. 127
To date, randomized controlled trials (RCTs) have not demonstrated the superiority of any investigational therapy over TCS as first-line treatment for moderate to severe cGVHD.128,129 This has left a gap in approved therapeutic options, with no FDA-approved steroid-free regimen available as first-line for newly diagnosed cGVHD. 130 Given the heterogeneity and progressive nature of cGVHD, there is growing consensus that corticosteroids should not be considered the universal gold standard, and prospective clinical trials exploring novel, steroid-free strategies are urgently needed to improve long-term patient outcomes.129,130
Prospects in Topical Therapies
Emerging topical treatments for cutaneous cGVHD aim to address the limitations of existing therapies while reducing systemic immunosuppressive exposure. A recent study explored the use of topical 5-fluorouracil (5-FU) cream for cases of cGVHD localized to chronically photodamaged skin, hypothesizing that reducing keratinocyte mutational burden may lower aberrant immune responses. In this case series, patients demonstrated clinical improvement following treatment with 5-FU, suggesting a potential role for this antineoplastic agent in targeted cGVHD therapy. However, further prospective trials are necessary to confirm its efficacy and safety in larger patient cohorts. 131
Interim results from a randomized, double-blind, vehicle-controlled phase II study of ruxolitinib 1.5% cream in patients with nonsclerotic and superficially sclerotic cGVHD demonstrated reductions in involved BSA and lesion severity compared with vehicle. 132 Treatment was generally well tolerated, and no serious treatment-related adverse events were reported. As these findings are based on interim analyses, longer-term follow-up and confirmatory studies are needed to establish durability of response and safety.
Reports of topical ruxolitinib/delgocitinib use in superficially sclerotic genital dermatoses, including lichen sclerosus, have described symptomatic improvement.133,134 However, these findings derive from small case series and are not specific to cutaneous cGVHD; therefore, extrapolation to broader cutaneous GVHD populations should be made cautiously.
Overall, topical Janus kinase (JAK) inhibition represents a promising investigational approach for localized, steroid-refractory cutaneous cGVHD, but further randomized studies are required to define its role in clinical practice.
Field Therapies
Phototherapy
For skin conditions involving >15% BSA, it may be difficult to apply topical therapies, making phototherapy indispensable. Phototherapy in cutaneous cGVHD exerts its effect through localized immune modulation. Ultraviolet (UV) light induces apoptosis of activated T cells and suppresses skin-resident APCs, such as Langerhans cells and dermal dendritic cells, thereby reducing local inflammation and promoting antigen tolerance. This process downregulates pathogenic T cell responses and shifts the cutaneous immune milieu toward regulatory and tolerogenic pathways, without causing systemic immunosuppression. Additionally, phototherapy can increase regulatory T cells (Foxp3+), further supporting immune tolerance in the skin.135-137 Over time, this process promotes antigen tolerance without inducing widespread immunosuppression. Different types of phototherapies include PUVA (psoralen plus UVA, 320-400 nm), UVA1 (340-400 nm), broad-band (280-320 nm) and narrow-band UVB (NB-UVB) (311-313 nm), each distinguished by their wavelengths, depth of skin penetration, and carcinogenesis risk (Supplemental Table 2). 85 As a non-systemic treatment, phototherapy is particularly beneficial for patients who require immune modulation but may not tolerate additional systemic immunosuppression. Since phototherapy services are available in many outpatient settings, access is often feasible for patients based on geographic location and center availability. Patients can also purchase home units and handheld devices (latter costing <200USD/270CAD) that are easy to operate. However, analyses of the effectiveness of different phototherapy approaches through RCTs in cutaneous cGVHD are lacking. 85
NB-UVB
NB-UVB phototherapy has emerged as an effective, skin-directed treatment for cutaneous cGVHD, particularly in patients who do not adequately respond to systemic immunosuppressive therapies. NB-UVB delivers light at a narrow wavelength range without requiring a photosensitizer, thereby reducing the risk of systemic side effects and long-term toxicities associated with psoralen-based regimens. Multiple studies demonstrated NB-UVB to be non-carcinogenic in long-term use, unlike Broad Band UVB (BB-UVB).138-140
Clinical evidence supports its efficacy in cGVHD. In a retrospective study by Ballester-Sánchez et al, 141 of 16 patients with cutaneous cGVHD treated with phototherapy, 6 received NB-UVB and achieved significant clinical improvement, with patients able to reduce their topical and systemic corticosteroid doses. Similarly, Enk et al 142 evaluated NB-UVB in patients with cGVHD who were refractory to standard immunosuppressive treatment; they found that NB-UVB produced complete clearance of lesions in patients with lichenoid/papulosquamous variant, while those with sclerodermoid changes experienced marked relief of pruritus despite less pronounced improvements in skin induration.
Even in pediatric patients, Brazzelli et al 143 reported a complete response in 80% of cases after a median of 29 treatment sessions over approximately 7.5 weeks, underscoring NB-UVB’s robust efficacy and rapid action in cGVHD. George et al 144 further supported these findings in an Indian cohort of patients, reporting high response rates among individuals with predominant skin involvement, which establishes NB-UVB as a valuable option for those refractory to standard therapies.
Balighi et al conducted a case series study in which 7 patients with mucocutaneous cGVHD were treated with NB-UVB phototherapy. They categorized the patients based on the type of skin lesions (lichenoid, sclerodermoid, or mixed) and documented that overall BSA involvement was significantly reduced by the end of treatment. The lichenoid group showed the most rapid and marked improvement (a decrease from 51% to 16.6%, a 53.3% ORR), whereas the sclerodermoid group experienced moderate improvement, due to limited UVB penetration into the deeper dermis. Importantly, no serious adverse effects were reported, and patients also benefited from improved quality of life and reduced doses of concurrent immunosuppressive drugs. 145
A recent systematic review by Fachler-Sharp et al 113 consolidates the evidence from multiple studies and reports that NB-UVB phototherapy achieves an overall response rate (ORR) of 94% in cutaneous cGVHD, with patients undergoing an average of 26 treatment sessions and experiencing only mild adverse effects in 8.6% of cases.
PUVA
PUVA has shown promise in improving deeper-seated skin lesions while offering a steroid-sparing effect. In PUVA therapy, 8-methoxypsoralen (8-MOP) is administered (systemically or in a bath, topically) prior to controlled UVA exposure, leading to its activation and subsequent intercalation into the DNA of activated lymphocytes. This results in the formation of crosslinks that induce apoptosis, thereby reducing the inflammatory infiltrate and modulating antigen presentation by Langerhans cells. These immunomodulatory effects help to alter the local cytokine environment that drives the chronic inflammatory process in cGVHD.146,147 Hand and foot PUVA is also available where 1% methoxsalen lotion is diluted to a final concentration of 0.1% in a cleanser (not cream) and applied to hands and feet 20 minutes prior to exposure to UVA light. Subsequently, the “methoxsalen cleanser” is easily washed off therefore limiting adverse events should the patient expose hands or feet to the sun afterwards.
Clinical evidence supports the use of PUVA in cutaneous cGVHD, especially for sclerotic forms of the disease. Ballester-Sánchez et al 141 retrospectively reviewed a series of 16 patients treated predominantly with PUVA, reporting complete clearance in 9 patients and partial improvement in 7, with many patients able to reduce their systemic corticosteroid dosages. Jampel et al 148 demonstrated that, in patients with the lichenoid variant of cGVHD, PUVA induced significant clinical and histologic improvements in most cases. Creamer et al 149 extended these findings by describing an eczematoid variant of cGVHD that, despite its aggressive presentation, responded well to PUVA with marked reductions in lesion severity and pruritus.
Leiter et al 150 reported that bath PUVA can be an effective adjunct in patients with extensive cutaneous cGVHD, allowing for significant tapering of systemic corticosteroids.
Fachler-Sharp et al 113 further consolidates these findings, the review encompassed 28 studies, reporting that among patients treated with PUVA, the ORR was approximately 89.9% after a mean of 33 treatments, with adverse events recorded in about 54% of cases (mostly mild).
It not only leads to significant clinical and histologic improvements but also reduces the systemic immunosuppressive burden—a critical factor in the long-term management of these patients. PUVA therapy, while effective, is the most carcinogenic of all phototherapy options. Critically, use of PUVA is limited to <200 treatments or <2000 J/cm2 in a lifetime to avoid development of melanoma and keratinocyte carcinoma.140,151-153
UVA1
UVA1 phototherapy has emerged as a promising treatment modality for patients with cutaneous cGVHD, particularly those with sclerodermoid changes that are often refractory to conventional immunosuppressive therapy. The therapeutic benefits of UVA1 are thought to stem from its deeper dermal penetration, which facilitates modulation of fibroblast activity, upregulation of collagen-degrading matrix metalloproteinases, and induction of apoptosis in pathogenic T lymphocytes. UVA1 is significantly less carcinogenic than PUVA and is therefore considered safe and suitable for use in pediatric patients.154,155
Connolly et al 156 demonstrated that in a heterogeneous cohort including 25 patients with cGVHD good therapeutic efficacy was achieved with UVA1 phototherapy, with a clear dose–response relationship showing that higher doses (80-120 J/cm2) yielded better clinical improvement compared to low- or medium-dose regimens. In a similar vein, Wetzig et al 157 reported that medium-dose UVA1 therapy resulted in complete responses in 60% and partial responses in 30% of cGVHD patients, with improvements sustained over a median follow-up of 14 months.
Notably, emerging evidence points to a differential response based on the subtype of cutaneous cGVHD. One study indicated that UVA1 might be particularly effective for the sclerotic type of cGVHD due to deeper penetration of light into tissue. In that study, 3/5 patients with sclerotic disease achieved complete remission, and the remaining 2 experienced partial improvement after an average of 21 treatment sessions. In contrast, patients with the lichenoid variant relapsed within a month after discontinuing treatment and required extended maintenance therapy to sustain clinical benefit. 158
Objective measurements provide further support for the clinical benefits of UVA1. Osmola et al used high-frequency ultrasonography and cutometer methods to assess changes in skin elasticity and dermal thickness in sclerodermoid cGVHD patients treated with medium-dose UVA1. Their results showed significant improvements in skin texture and elasticity, corroborating clinical assessments of reduced sclerosis. 159 Complementary imaging studies, such as those by Gottlöber et al, 160 have illustrated that reduction in skin thickness, as measured by 20 MHz sonography, correlates with clinical improvement in GVHD lesions.
In a comprehensive review of phototherapy for sclerosing skin conditions, Teske and Jacobe (2016) underscored the unique advantages of UVA1 therapy. They highlighted that UVA1 not only increases collagenase activity (thereby promoting collagen degradation) but also exerts immunomodulatory effects that are crucial for reducing fibrotic processes in cGVHD. 161 These mechanistic insights are further supported by reports from Schlaak et al, 162 who, although focusing on acute cutaneous GVHD, observed that UVA1 allowed for tapering of systemic immunosuppression, a benefit that may extend to the cGVHD setting.
Case reports have also highlighted the potential of low-dose UVA1. Grundmann-Kollmann et al described a patient with sclerodermic cGVHD refractory to conventional immunosuppression who responded favorably to a low-dose regimen (20 J/cm2 administered 4 times per week). The patient experienced marked softening of sclerotic lesions and improved joint mobility, suggesting that even lower doses of UVA1 can be effective in selected cases. 163 A related report by Ständer et al 164 in a small case series confirmed that UVA1, using doses ranging from 20 to 50 J/cm2, yielded significant clinical improvements, although the optimal dosing may depend on individual patient factors.
Fachler-Sharp et al further support these observations. A review, which included 6 studies aforementioned above encompassing 132 patients treated with UVA1, reported an ORR of 89.3% with a mean of approximately 26 treatments. The review also highlighted that while adverse events were relatively frequent (around 70%), they were predominantly mild manifesting as reversible tanning, mild erythema, and other transient skin changes that rarely necessitated treatment discontinuation. 113 Importantly, UVA1 carries a very low risk of carcinogenesis unlike PUVA or BB-UVB
Its ability to modulate collagen metabolism and immune responses provides a rational basis for its use, especially in patients who are refractory to or cannot tolerate systemic immunosuppressive therapies. While medium- to high-dose regimens appear to be particularly beneficial, further prospective studies are needed to optimize dosing protocols and assess long-term outcomes. UVA1 and PUVA therapies also have drawbacks such as very long treatment times, paucity of sites offering, requiring frequent clinic visits/travel, high equipment costs, need for eye protection, risk of phototoxic reactions, gastrointestinal upset with oral psoralen, increased risk of skin cancers (particularly melanoma and squamous cell carcinoma with PUVA), photoaging, contraindications in pregnancy, and need for lifelong cancer surveillance.
Extracorporeal Photopheresis
ECP has been established as a key second-line therapy for steroid-resistant (SR) cGVHD, demonstrating consistent, progressive skin improvement across diverse clinical settings. ECP involves leukapheresis, ex vivo exposure of mononuclear cells to 8-MOP and UVA, and reinfusion, resulting in immunomodulation (without immunosuppression) through induction of regulatory T cells, apoptosis of pathogenic lymphocytes, and modulation of dendritic cell function, without causing global immunosuppression or increasing infection risk. ECP is especially valuable for patients with widespread or sclerotic skin involvement where topical therapies (TCS, TCIs, topical JAK inhibitors) and phototherapy (NB-UVB, PUVA, UVA1) are insufficient or impractical.165,166
In their 2020 review, Drexler et al 167 reported that pooled response rates for cutaneous cGVHD reached 74% (complete or partial) after prolonged ECP courses, highlighting both rapid onset, often within the first 8 to 12 weeks, and durable benefit with minimal toxicity. The only multicenter, randomized phase 2 study by Flowers et al compared ECP plus standard immunosuppression versus immunosuppression alone in 95 patients with cutaneous cGVHD; at week 12 the ECP arm achieved a 40% overall skin response rate versus 10% in controls (P = .002), alongside a ≥50% steroid-dose reduction in 17% of patients (vs none in controls), without serious adverse events. 168
Complementing these RCT data, a prospective crossover trial of 29 patients by Greinix et al demonstrated that cutaneous response rates rose from 8% on conventional therapy to 31% during ECP (P = .04), with accompanying improvements in lesion severity scores and steroid-sparing in one-third of participants. In a single-center prospective series, Foss et al treated 25 SR-cGVHD patients with ECP and reported regression of skin pathology in 80% of cases, with oral-mucosal and joint improvements. Later, Dignan et al 169 observed ORRs of 72% in lichenoid and 80% in sclerodermatoid skin variants at 6 months among 27 patients, achieving corticosteroid reductions in nearly 90% of responders.
Retrospective data further reinforce these benefits: Del Fante et al 170 found an 80.5% overall skin response rate in 102 patients treated with ECP plus immunosuppression, with a median prednisone dose reduction of 77.6% from baseline. More recently, Jagasia et al 171 conducted a randomized pilot of first-line ECP added to standard of care in 53 patients and reported a higher, though not statistically powered, skin ORR of 74.1% versus 60.9%, while preserving quality of life in the ECP arm. Across case series summarized by Canto and colleagues, median cutaneous response rates of 75% and steroid-tapering in approximately 65% of patients underscore ECP’s real-world effectiveness and tolerability (no serious ECP-related adverse events reported). 172
The role of ECP in up front and combination therapy requires further investigation and multiple clinical trials are ongoing to evaluate this. Additionally, it must be mentioned that this therapy poses practical challenges for patients—the frequency, cost and travel distance to an ECP site are all barriers to care with ECP.
Systemic Therapies
Systemic corticosteroids remain the cornerstone of first-line therapy for NIH-defined moderate to severe cGVHD, including its cutaneous manifestations. They are typically initiated at a prednisone-equivalent dose of 0.5 to 1 mg/kg/day, often combined with a CNI such as cyclosporine or tacrolimus or a JAK inhibitor (as detailed later). 173 This approach is consistent with the recently published Cell Therapy Transplant Canada Consensus-Based Guideline, which provides the first nationwide recommendations for diagnosis and treatment of cGVHD and emphasizes standardized systemic management across Canadian centers. 87
Despite broad anti-inflammatory effects, corticosteroids provide only partial and often temporary control. In a large single-center study by Inamoto et al, the 12-month failure-free survival (FFS) following initial systemic therapy, which included corticosteroids, was only 54%. Nearly half of patients either required second-line/another systemic treatment, experienced non-relapse mortality, or had recurrent malignancy during initial therapy. 173 Notably, high initial doses did not correlate with improved outcomes.
Real-world data further emphasize the limitations of corticosteroids. In a claim-based analysis by Bachier et al, corticosteroids were the most frequently prescribed therapy in first-, second-, and third-line settings. Yet over 70% of patients progressed beyond first-line treatment, and 47% required a third-line, illustrating the frequent need for additional agents and the lack of durable control with systemic corticosteroids alone. 4 Additionally, the chronic use of steroids contributes significantly to morbidity, particularly in patients with skin involvement, where steroid-induced myopathy, skin thinning, and delayed wound healing compound disease-related impairment. They are associated with a high burden of toxicity and suboptimal long-term efficacy. The combined inflammatory and fibrotic pathophysiology likely explains limited steroid responsiveness in fibrotic skin disease. The development of fibrosis in cGVHD involves macrophage infiltration, Th17 signaling, and fibroblast activation, processes that are not easily addressed by steroids alone. 174
Recent therapeutic approaches for cGVHD have transitioned from broad immunosuppressive regimens relying heavily on prolonged, high-dose corticosteroids to more targeted strategies directed at specific immune pathways implicated in disease mechanisms (Table 3, Figure 2). These emerging therapies selectively target donor-derived alloreactive T lymphocytes, auto- and alloreactive B lymphocytes, as well as CD4+ FoxP3+ regulatory T cells. 175 Significant progress in understanding cGVHD pathogenesis, alongside improved diagnostic guidelines, refined disease severity grading, clearer response criteria, and standardized clinical trial endpoints, has culminated in the FDA’s approval of multiple agents for patients requiring second-line therapy or beyond.
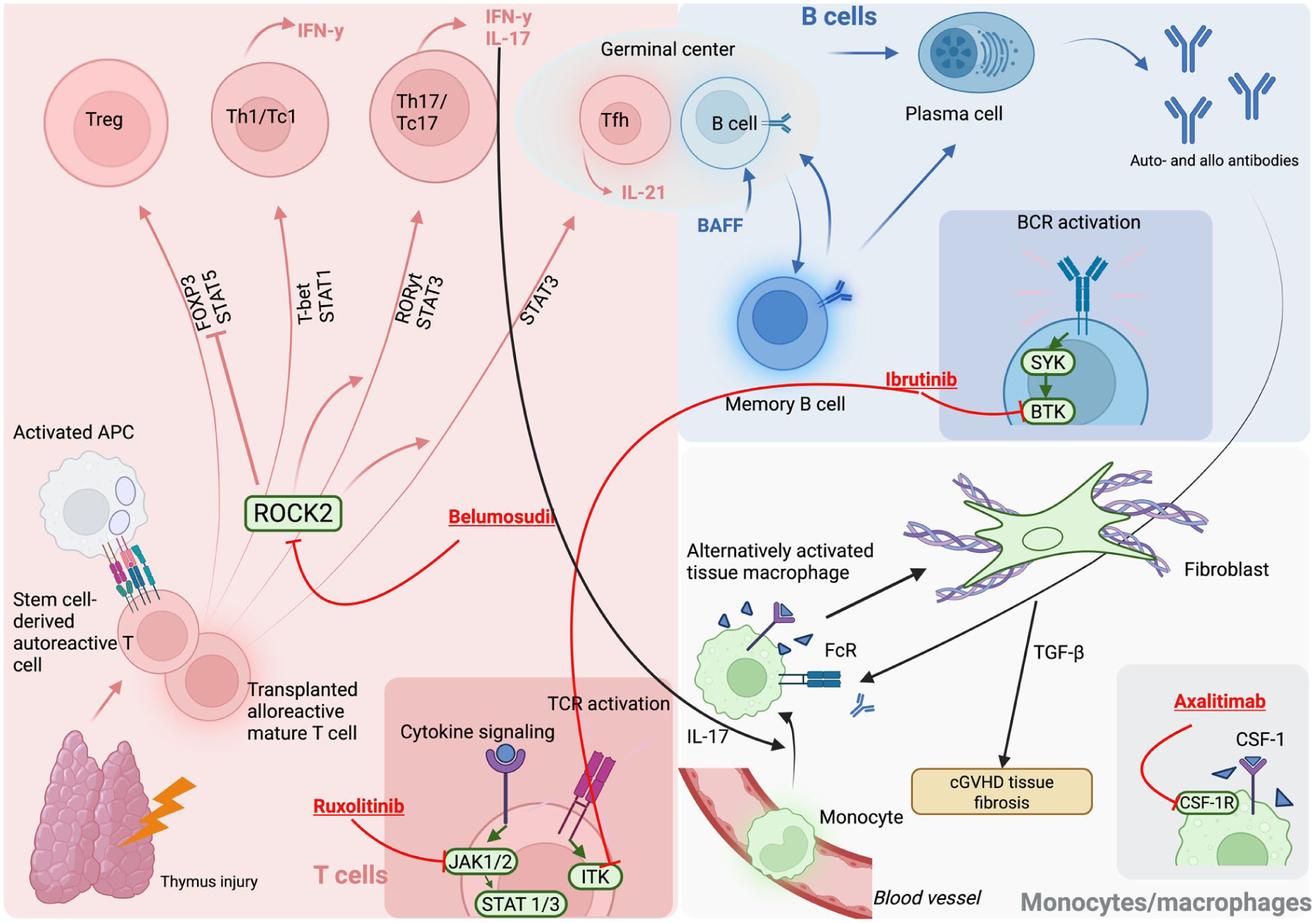
Integrated cellular and molecular mechanisms underlying cGVHD pathogenesis and therapeutic targets. The figure illustrates the complex interplay between donor-derived immune cells and tissue-resident effectors that drive the pathogenesis of cGVHD. Thymic injury impairs central tolerance and permits the emergence of autoreactive donor T cells that become activated upon encountering host APCs. Activated APCs promote differentiation of Th and cytotoxic (Tc) subsets through cytokine and TCR signaling cascades involving JAK1/2–STAT1/3 and ITK pathways. Ruxolitinib, a JAK1/2 inhibitor, attenuates this signaling, thereby reducing effector T-cell activation. Activated donor T cells polarize toward Th1/Tc1 (T-bet, STAT1) and Th17/Tc17 (RORγt, STAT3) lineages, producing IFN-γ and IL-17, which perpetuate tissue inflammation. ROCK2 signaling contributes to Th17/Tc17 differentiation and suppression of Tregs through inhibition of FOXP3/STAT5; blockade by belumosudil restores immune balance. Tfh cells within germinal centers secrete IL-21, driving B-cell maturation and differentiation into plasma cells that produce auto- and alloantibodies. Elevated BAFF supports survival and activation of memory B cells through BCR signaling mediated by BTK and SYK kinases, which are inhibited by ibrutinib. Circulating monocytes differentiate into alternatively activated tissue macrophages, which interact with immune complexes through Fc receptors (FcR) and secrete IL-17 and TGF-β, promoting fibroblast activation and tissue fibrosis, the pathological hallmark of cGVHD. The CSF-1/CSF-1R axis sustains macrophage survival and differentiation; axatilimab, a monoclonal antibody targeting CSF-1R, interrupts this profibrotic signaling loop. Together, these interconnected immune and stromal pathways link defective thymic tolerance, sustained T- and B-cell activation, and macrophage-driven fibrogenesis in the chronic phase of GVHD, highlighting multiple therapeutic targets currently under clinical investigation. APC, antigen-presenting cell; BAFF, B-cell activating factor; BCR, B-cell receptor; BTK, Bruton’s tyrosine kinase; CSF-1(R), colony-stimulating factor 1 (receptor); IFN-γ, interferon-gamma; IL, interleukin; ITK, IL-2-inducible T-cell kinase; JAK, Janus kinase; ROCK2, Rho-associated coiled-coil kinase 2; STAT, signal transducer and activator of transcription; TCR, T-cell receptor; Tfh, T follicular helper cell; Th, T helper cell; Treg, regulatory T cell; TGF-β, transforming growth factor-beta, cGVHD, chronic graft-versus-host disease.
Ibrutinib
Ibrutinib, a first-in-class irreversible inhibitor of Bruton’s tyrosine kinase and interleukin-2 inducible kinase, has emerged as an important therapeutic advancement in the management of cGVHD following failure of corticosteroid therapy. Its dual inhibition capability makes it uniquely suited to address both B- and T-cell-mediated pathophysiological pathways involved in cGVHD development and persistence. 176
The pivotal phase 1b/2 study conducted by Miklos et al provided clinical confirmation of ibrutinib’s efficacy in patients with cGVHD resistant to corticosteroids. In this open-label trial, patients received ibrutinib 420 mg daily following failure of 1 to 3 prior systemic therapies. At a median follow-up of 13.9 months, the ORR was 67%, with sustained responses in 71% of responders for at least 20 weeks. Additionally, there was a notable reduction in corticosteroid doses required by responding patients, with some patients entirely discontinuing corticosteroids. 116
Extended follow-up from this same trial by Waller et al at a median of 26 months confirmed these initial findings, demonstrating a sustained ORR of 69%, including complete responses (CRs) in 31% of patients. Multi-organ responses were common, and significant improvements were reported in sclerotic manifestations, with 61% of patients exhibiting a reduction in fibrosis. Importantly, corticosteroid dose reduction continued over the longer follow-up, further highlighting ibrutinib’s corticosteroid-sparing potential. Despite a notable rate of adverse events (primarily fatigue, diarrhea, and infections), the study supported ibrutinib’s acceptable safety profile for long-term administration. 177
Ruxolitinib
Ruxolitinib, an oral inhibitor of JAK1/JAK2, has demonstrated significant efficacy in the treatment of SR-cGVHD. Ruxolitinib’s mechanism involves suppressing the inflammatory pathways central to cGVHD pathogenesis, making it a promising therapeutic option for patients who have not responded to initial corticosteroid therapy.
The REACH3 trial, a pivotal phase 3 randomized study, confirmed ruxolitinib’s efficacy compared to the best available therapy (BAT), which commonly included ECP. Patients treated with ruxolitinib had a significantly higher ORR at 24 weeks (49.7%) compared to those receiving BAT (25.6%) and also exhibited longer median FFS (>18.6 months vs 5.7 months). Additionally, symptom response based on the modified LSS was significantly better in the ruxolitinib group (24.2% vs 11.0%). 121
Real-world studies have further substantiated ruxolitinib’s therapeutic value. A multicenter retrospective study involving heavily pretreated patients revealed ORRs of approximately 49% to 55% at 3 and 6 months, respectively, and enabled significant corticosteroid dose reduction. At 12 months, about 38% of patients could discontinue prednisone completely, highlighting its corticosteroid-sparing benefit. Notably, severe cGVHD and high comorbidity index scores correlated with poorer outcomes, suggesting careful patient selection is crucial. 178
A meta-analysis conducted by Fan et al, encompassing multiple studies and 1580 patients, confirmed high ORRs (78%) with ruxolitinib treatment, though CRs were less frequent (15%). Response rates varied significantly by organ involvement, with oral/mucosal and cutaneous cGVHD showing the highest responses. The analysis underscored manageable adverse events, predominantly infections and cytopenias, which were comparable between pediatric and adult populations. 179
Further real-world data from Redondo et al in 48 SR-cGVHD patients demonstrated an impressive ORR of 77%, including 15% achieving CR. Importantly, 54% of patients successfully tapered steroids, with complete discontinuation in 21%. Responders had notably better survival rates compared to non-responders, reinforcing the clinical benefit and tolerability of ruxolitinib. 180
Ferreira et al provided long-term follow-up data supporting sustained efficacy, reporting an ORR of 89%, with a significant proportion of patients achieving complete remission (26%). Long-term data also emphasized the durability of responses, with >50% of patients being still in remission and steroid-free at extended follow-up periods, indicating that ruxolitinib not only achieves responses but also sustains them. 181
Wu et al corroborated these findings, reporting a 70.7% ORR among 41 patients, with meaningful responses even in heavily pretreated patients. Lung involvement and matched related donors were identified as factors associated with less favorable responses. Major adverse effects observed included cytopenias and infections, consistent with findings from other studies, and reinforcing the need for careful patient monitoring and supportive care. 182
Notably, in the Novitzky-Basso cohort, 75.7 % of ruxolitinib-treated patients had active cutaneous involvement at baseline, underscoring the high burden of skin disease in this refractory population. This severity did not blunt ruxolitinib’s benefit: by month 6, 24.1 % of patients in the ruxolitinib arm had completely discontinued prednisone compared to just 2.0 % of matched historical controls, a 22.1 % absolute difference that closely parallels sustained cutaneous control and symptom resolution. In the propensity-matched subgroup (65.9 % baseline skin involvement in the ruxolitinib group vs 54.5 % in controls), ruxolitinib still led to a 22.2 % greater rate of steroid discontinuation by 6 months, in concert with its superior 12-month FFS of 74.7 % versus 19.1 % (P < .001). Together, these skin-specific outcomes, high baseline involvement, rapid and durable steroid tapering, and exceptional FFS, reinforce ruxolitinib’s real-world efficacy in controlling cutaneous cGVHD. 183
Belumosudil
Belumosudil, a selective oral inhibitor of rho-associated coiled-coil kinase 2 (ROCK2), is approved for the treatment of cGVHD, particularly following failure of corticosteroid therapy and other systemic treatments. ROCK2 inhibition modulates immune dysregulation and fibrosis by shifting T cell polarization from pro-inflammatory Th17 cells toward regulatory T cells and reducing fibrosis through inhibition of profibrotic signaling pathways.
The pivotal ROCKstar study evaluated belumosudil’s efficacy in heavily pretreated patients with cGVHD who had received 2 to 5 prior systemic therapies. This randomized phase 2 trial reported ORRs of 74% and 77% with belumosudil 200 mg daily and twice daily, respectively. Organ-specific analyses of the ROCKstar trial revealed that cutaneous involvement had the best ORR of 37%. Among the 41 subjects who achieved a skin response, 11 (27%) exhibited a decrease in sclerotic features, 15 (37%) showed a reduction in BSA involvement, and 13 (32%) demonstrated improvements in both sclerosis and BSA involvement. The median time to skin response was 5 weeks (range, 4-66) and 91% of responses occurred within 6 months. The median duration of response in skin responders was 54 weeks, and 59% of these patients maintained their improvement for at least 20 weeks. Concurrently, 65% of all subjects reduced their corticosteroid dose (mean reduction 45%), and 21% were able to discontinue steroids entirely by 12 months, indicating corticosteroid dose reductions during treatment. The drug was also associated with only 12% discontinuation rate due to adverse events. 122
Further data were reported in another phase IIa dose-finding trial by Jagasia et al, which showed an ORR of 65% across multiple dosage regimens. The median response duration was approximately 35 weeks, and belumosudil facilitated substantial corticosteroid dose reductions and improvements in patient quality of life. Importantly, the FFS rate was 76% at 6 months, with no unexpected safety signals reported. 184
Tissue-level analyses from a ROCKstar companion study further demonstrated that ROCK2 inhibition directly affects effector sites, including oral mucosa and skin. 185 In oral mucosal biopsies, belumosudil significantly reduced collagen types I and III, decreased IL-17-producing cells, predominantly from non-T-cell sources, and reduced macrophage infiltration, with macrophage reduction correlating with lower collagen deposition. Additionally, salivary TGF-β1 levels declined following treatment, supporting a local antifibrotic effect. 185 These findings provide mechanistic evidence that belumosudil mitigates inflammation and fibrosis directly within cGVHD target tissues.
Real-world data have provided further information regarding response rates and safety. Modi et al reported a lower ORR (46.7%) at a single institution, likely due to a higher proportion of patients previously treated with ruxolitinib or concurrent therapy with ruxolitinib. This experience highlighted a significant risk of infections, particularly when used in combination with ruxolitinib, emphasizing the need for cautious patient monitoring. 186
A German-Swiss multicenter retrospective analysis by Heidenreich et al reported an ORR of 42% in a heavily pretreated cohort and a median FFS of 16.5 months. Despite being heavily pretreated, the safety profile was consistent with prior reports, though infections were noted as significant adverse events. 187
Similarly, a French multicenter retrospective analysis by Michonneau et al reported an ORR of 57.3%, with significant responses in liver and oral cGVHD. The median FFS was favorable, with low discontinuation rates due to adverse events, consistent with previously described safety data. 188
Petty et al focused specifically on belumosudil’s role as a steroid-sparing agent in cutaneous cGVHD. They demonstrated substantial reductions in corticosteroid dosage, with significant decreases in the percentage of BSA involved in patients with cutaneous involvement. 189
Axatilimab
Axatilimab is a humanized IgG4 monoclonal antibody that selectively targets the colony-stimulating factor 1 receptor (CSF-1R), a key regulator of macrophage survival, proliferation, and differentiation. Given the central role of profibrotic macrophages in cGVHD pathogenesis, particularly in driving fibrosis and sustained inflammation, axatilimab has emerged as a therapeutic strategy for patients with advanced or refractory cGVHD.
The AGAVE-201 trial, a pivotal phase II multinational randomized study, evaluated axatilimab at 3 dosing regimens (0.3 mg/kg every 2 weeks, 1 mg/kg every 2 weeks, and 3 mg/kg every 4 weeks) in 241 patients with recurrent or refractory cGVHD leading to FDA approval. The primary endpoint, ORR within the first 6 cycles, was met in all dosing groups: 74% in the 0.3 mg/kg group, 67% in the 1 mg/kg group, and 50% in the 3 mg/kg group. Additionally, a clinically significant reduction in symptom burden (>5-point drop in the modified LSS) was reported in 60%, 69%, and 41% of patients, respectively. In patients with sclerotic skin manifestations, which constituted over 90% of the cohort, reduction in sclerotic skin surface area was observed in 44% of those receiving 0.3 mg/kg every 2 weeks, 34% of those on 1 mg/kg every 2 weeks, and 60% of those on 3 mg/kg every 4 weeks. Correspondingly, improvements in skin sclerosis were reported in 66%, 56%, and 60% of patients across these dose levels, respectively. These objective measures were mirrored by patient-reported symptom relief: 73%, 77%, and 68% of participants in the 0.3 mg/kg, 1 mg/kg, and 3 mg/kg arms, respectively, experienced a clinically meaningful reduction in skin-thickening symptoms on the modified LSS. 123
The median time to response was under 2 months and 91% of cutaneous improvements occurring within 6 months. Many patients experienced rapid corticosteroid tapering or discontinuation. Twelve-month overall survival was high across all groups, exceeding 80%, with a 12-month FFS ranging from 44% to 59%. Axatilimab-related adverse events were generally dose-dependent and consistent with CSF-1R inhibition but rarely resulted in treatment discontinuation, especially at the 0.3 mg/kg dose. 123
Building on these findings, the phase I/II open-label study by Kitko et al evaluated axatilimab in 40 patients with active cGVHD after failure of at least 2 prior lines of systemic therapy. The study identified 3 mg/kg every 4 weeks as the optimal biologic dose, with a recommended phase II dose (RP2D) of 1 mg/kg every 2 weeks based on favorable safety, pharmacodynamic activity, and response outcomes. Among evaluable patients, the ORR was 67%, with responses across all organs, including skin and lung involvement. In the phase II cohort, the ORR at cycle 7 day 1 was 50%, but when considering best responses within the first 6 cycles, the ORR increased to 82%. At baseline, 35 of the 40 patients (87.5%) enrolled in the Kitko et al study had active cutaneous involvement (77.5% with a skin features score ≥2), reflecting the heavy fibrotic burden in this cohort. Although only 5 of these 35 patients (14%) achieved a formal partial or complete skin response by NIH 2014 criteria during treatment, the majority experienced meaningful improvements in fibrotic manifestations: among the 31 patients with sclerotic skin disease, 26 (84%) showed investigator-rated decreases in skin tightening and reported symptom relief on the LSS. Responses were rapid, with a median time to first documented skin improvement of 5 weeks (range 4-20), and durable, as many patients maintained reduced induration and greater range of motion through 20 weeks of follow-up. These findings underscore axatilimab’s capacity to ameliorate both clinical and patient-reported measures of cutaneous sclerosis in refractory cGVHD, even when formal response rates appear modest. Axatilimab also demonstrated a favorable safety profile with most adverse events being mild and on-target (eg, liver enzyme elevations, fatigue, periorbital edema) and no cytomegalovirus reactivations or invasive fungal infections were observed. 190 Taken together, these organ-specific analyses underscore axatilimab’s capacity to both diminish fibrotic skin involvement and alleviate patient-reported symptom burden in refractory cGVHD.
Prospects in Systemic Therapies
Multiple clinical trials are under way to evaluate novel therapies in cGVHD (Table 5).
Ongoing Clinical Trials in Cutaneous cGVHD.
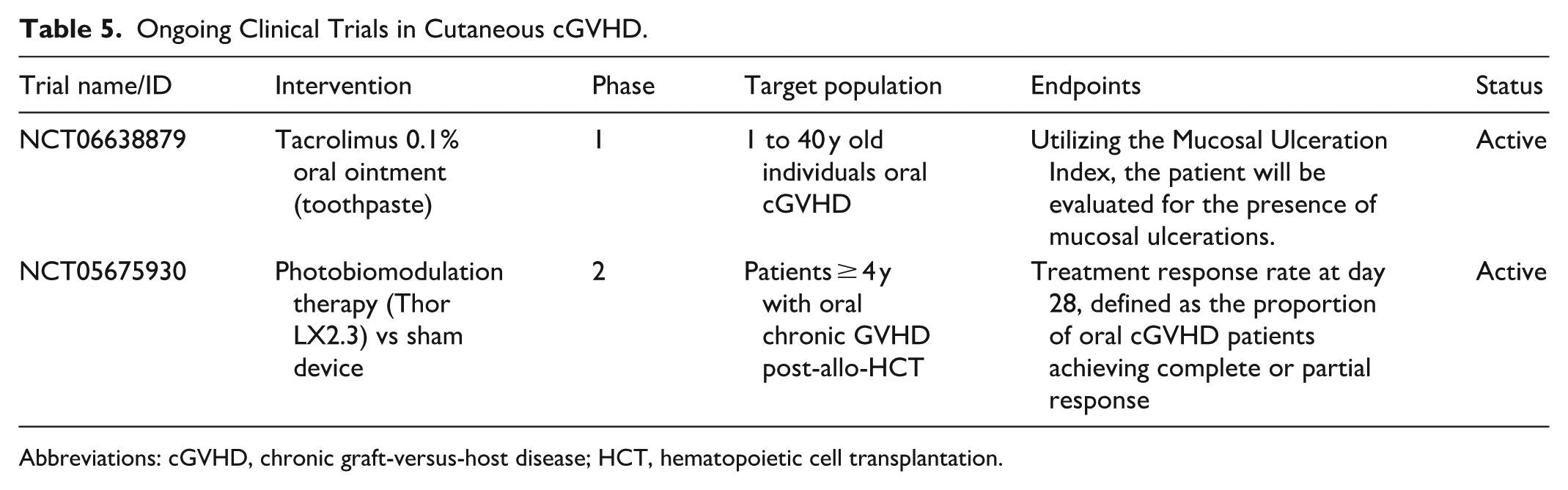
Abbreviations: cGVHD, chronic graft-versus-host disease; HCT, hematopoietic cell transplantation.
Rovadicitinib
Rovadicitinib is a first-in-class, oral dual inhibitor of JAK1/2 and ROCK1/2 developed to simultaneously target the inflammatory and fibrotic pathways implicated in cGVHD. This dual mechanism is particularly relevant in cutaneous cGVHD, where both immune-mediated damage and fibrotic remodeling contribute to disease persistence and resistance to therapy.
In a phase 1b/2a multicenter study conducted in China (NCT04944043), rovadicitinib was evaluated in 44 patients with moderate-to-severe glucocorticoid-refractory or -dependent cGVHD. The majority of patients were heavily pretreated, with nearly two-thirds exhibiting involvement of at least 4 organs. Skin involvement was observed in 59.1% of patients, many of whom presented with sclerotic features, an especially challenging manifestation due to its fibrotic nature and limited responsiveness to existing therapies. 191
Rovadicitinib demonstrated encouraging clinical activity in the skin. Among the 26 patients with cutaneous involvement, the best ORR was 65.4% at 24 weeks. Notably, 57.6% of patients with evaluable BSA scores achieved a clinical response. Improvement was also seen in patients with skin sclerosis, as 50% of those presenting with NIH-defined sclerotic features experienced a reduction in severity scores. These findings suggest that rovadicitinib may address both inflammatory and fibrotic cutaneous manifestations, a therapeutic gap unmet by other agents such as ruxolitinib, which has shown limited efficacy in sclerotic skin disease. 191
The response in skin was often accompanied by a rapid onset of systemic benefit, with most responders achieving partial improvement within the first month of therapy. These clinical gains translated into tangible quality of life improvements, as measured by the LSS, and allowed for meaningful tapering of corticosteroids in the majority of patients. Overall, these results position rovadicitinib as a promising option for patients with cutaneous cGVHD, especially in cases where fibrosis predominates or other targeted therapies have failed. 191
Importantly, the safety profile of rovadicitinib was manageable, with no dose-limiting toxicities observed in the cutaneous subgroup and a lower rate of grade ≥3 anemia compared to JAK inhibitors like ruxolitinib. 191 Given the therapeutic challenge posed by sclerotic skin involvement in cGVHD, the dual inhibition strategy employed by rovadicitinib—targeting both the JAK/STAT and ROCK pathways—offers a novel and potentially more effective approach to disease modulation in this difficult-to-treat population. Rovadicitinib is currently undergoing a Phase II clinical trial for cGVHD in the United States.
Conclusion
Despite unprecedented advances in our understanding of cGVHD, it remains a challenging and highly morbid complication of allogeneic HCT. 192 Mechanistic studies have clarified the roles of dysregulated B- and T- cell immunity, profibrotic macrophages, and aberrant wound-healing responses, broadening the framework beyond simple alloimmunity. Four decades after corticosteroids were first adopted as frontline therapy, they remain the mainstay of treatment, often yielding suboptimal responses and exposing patients to long-term toxicity. For the substantial proportion of patients who develop steroid-refractory disease, multiple systemic therapies are typically required, yet many fail to achieve durable remission, particularly in the setting of fibrotic or sclerotic skin involvement.
The approval of novel agents such as ibrutinib, ruxolitinib, belumosudil, and axatilimab marks significant milestones in the therapeutic landscape. Each targets distinct immune or stromal pathways, reflecting the multifaceted immunopathogenesis of cGVHD. However, the comparable response rates observed across these agents, despite their distinct mechanisms of action, highlight that cGVHD represents not a single disease entity, but rather a syndrome resulting from diverse immunologic insults converging on common clinical manifestations. This heterogeneity is mirrored in the clinic, where presentations range from lichenoid/papulosquamous/poikiloderma rashes to deep fascial sclerosis and can be especially difficult to recognize in SOC, demanding careful examination and multidisciplinary evaluation. The challenge, then, is not only to expand our pharmacologic arsenal, but to refine how we use it as well as to develop rational, biomarker-driven treatment algorithms that match each patient to the therapy most likely to benefit them. Parallel progress in diagnostics, including advanced imaging, validated biomarkers, and PRO measures, promises to complement the NIH grading system and permit earlier, more precise monitoring of disease activity.
Cutaneous cGVHD, with its broad phenotypic variability and functional consequences, exemplifies the urgency of this mission. Therapies that address both inflammation and fibrosis, such as dual JAK/ROCK inhibitors, may offer hope for patients with sclerotic disease, a subgroup historically resistant to standard immunosuppression. At the same time, deeper exploration of resistance mechanisms, long-term safety profiles, and combination strategies is essential to optimize outcomes and reduce the physical and emotional toll on transplant survivors. Equally critical are supportive measures, including sun protection, rehabilitation to preserve mobility, and vigilant skin-cancer screening delivered within integrated transplant dermatology programs will improve survivorship and quality of life.
The future of cutaneous cGVHD management lies in a more personalized paradigm, 1 that integrates molecular diagnostics, clinical phenotype, and patient-centered outcomes to guide early, targeted, and less toxic interventions. As our collective understanding deepens, and the therapeutic toolkit grows, the goal remains clear: to move beyond symptom control toward true disease modification, and ultimately, a meaningful restoration of skin health and quality of life.
Supplemental Material
sj-docx-1-cms-10.1177_12034754261455768 – Supplemental material for Adult Cutaneous Chronic Graft-Versus-Host Disease: An In-Depth Review
Supplemental material, sj-docx-1-cms-10.1177_12034754261455768 for Adult Cutaneous Chronic Graft-Versus-Host Disease: An In-Depth Review by Arafat Atique, Meghan Kanou, Elena Netchiporouk, May Chergui, Gizelle Popradi and Ivan V. Litvinov in Journal of Cutaneous Medicine and Surgery
Supplemental Material
sj-docx-2-cms-10.1177_12034754261455768 – Supplemental material for Adult Cutaneous Chronic Graft-Versus-Host Disease: An In-Depth Review
Supplemental material, sj-docx-2-cms-10.1177_12034754261455768 for Adult Cutaneous Chronic Graft-Versus-Host Disease: An In-Depth Review by Arafat Atique, Meghan Kanou, Elena Netchiporouk, May Chergui, Gizelle Popradi and Ivan V. Litvinov in Journal of Cutaneous Medicine and Surgery
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Ivan Litvinov received honoraria or consulting fees from Janssen, Bausch, Galderma, Novartis, Pfizer, Sun Pharmaceutical Industries, Leo Pharma, Sanofi, Amgen, Therakos, Kyowa Kirin, Johnson & Johnson, and Actilion. Dr. Netchiporouk has been a speaker or consultant or has received investigator-initiated research funding from AbbVie, Arcutis, Bausch Health, Boehringer Ingelheim International, Bristol Myers Squibb, Galderma, Janssen, LEO Pharma, Medexus, Novartis Pharmaceuticals, Pfizer, Sanofi Genzyme, Sun Pharmaceuticals, Eli Lilly, and UCB. Dr. Gizelle Popradi reports a relationship with Amgen, AbbVie, BMS, Daiichi Sankyo, Gilead, Jazz, Kyowa Kirin, Mallinckrodt, Medexus, Merck, Novartis, Paladin, Pfizer, Sean Gen, Sanofi, Servier, Sobi, and Takeda that includes: consulting or advisory and speaking and lecture fees. Other authors declare no conflicts of interest.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.