
Abstract
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) clinically characterized by relapses and/or progressive disability in the majority of cases. In middle Europe its prevalence is estimated as about 1:1000. 1 Epidemiological and genetic data suggest both a relevant genetic 2 and environmental 3 impact on its pathogenesis. Depending on geographical background prevalence, duration of follow-up, gender or ascertainment strategies, in 5–26% of all cases another family member is also affected by MS. 4 – 6 In MS high risk regions monozygotic twins show concordance rates of up to 30%6,7 with a major contribution from female pairs, resulting in far exceeding those rates for dizygotic twins.6,7 An additional 13% of the other, clinically not affected monozygotic and 9% of the respective dizygotic twins indeed demonstrate asymptomatic central demyelination in brain MRI studies, 8 though the fewest seem stringently to convert into multiple sclerosis (denoted in Willer et al. 7 ) In general, the closer the relationship between family members, the higher is the risk of disease manifestation. 6 However, to transfer this epidemiological issue into definite gene associations even in regions of tight linkage disequilibrium sometimes caused confusion rather than clarification, because both the affected MS patients and their unaffected relatives carry susceptible alleles, concomitantly. Recently, epistasis among MS susceptibility haplotypes, especially of the HLA locus, provided evidence to determine not only the development of the disease 9 but eventually also the disability outcome. 10
Clinically, the individual disease evolution is modestly predictable and depends mainly on MRI data, disability evolution or relapse rate in the first years of the disease.11,12 Whether familial or genetic factors determine the clinical outcome in MS affected patients or not is an issue of ongoing discussion. 13 – 15 This study focuses prospectively on epidemiological six-year follow-up data in familial MS in Germany. We here provide evidence for a comparatively strong within-sibling pair concordance for disease progression, and significantly higher familial risks for multifocal onset and conversion to secondary progression of familial MS.
Patients and methods
Patients and their controls
One-hundred and eighty adult patients with familial MS were recruited from the Marburg University MS database, containing sociodemographic and clinical data of n = 943 MS patients (August 2010). All patients were diagnosed according to the McDonald criteria
16
and had at least one first or second degree relative with MS. Patients with a history of a concomitant disabling disease, affected but adopted relatives, refusal of participation of one of the affected relatives, or withdrawal of consent were excluded from the study. From all 156 evaluable familial cases, 98 siblings were included for the prospective follow-up analysis (Figure 1). Each affected sibling was carefully pair-matched for gender, age (±1 year), clinical course, disease duration (±6 months), and immunomodulating treatment (IFN-β 1a/1b vs. glatirameracetate vs. treatment naïve) with a patient from the remaining n = 763 non-familial sporadic cases (Table 1). All siblings were further stratified into pairs of sister/sister, sister/brother or brother/brother. To estimate the disease progression, n = 68 siblings (69.4%) and their matched controls were followed up prospectively each for a six-year period. All patients and controls gave written informed consent. The study was approved by the Institutional Review Board of Marburg University.
Flow-chart of analyses of n = 98 siblings with MS and their randomly matched sporadic controls. n = 68 (69.4%) were followed up prospectively for a six-year period. Clinical characteristics of n = 98 siblings with concomittant MS and their n = 98 controls of sporadic MS, randomly matched for age, gender, clinical course, disease duration and treatment at study entry Included n = 68 siblings and their matched controls. n = 44 of all included, n = 68 siblings and their matched controls. p < 0.018 (Kaplan–Meier analysis). RR: relapsing–remitting, SP: secondary-progressive, PP: primary-progressive, EDSS: Expanded Disability Status Scale.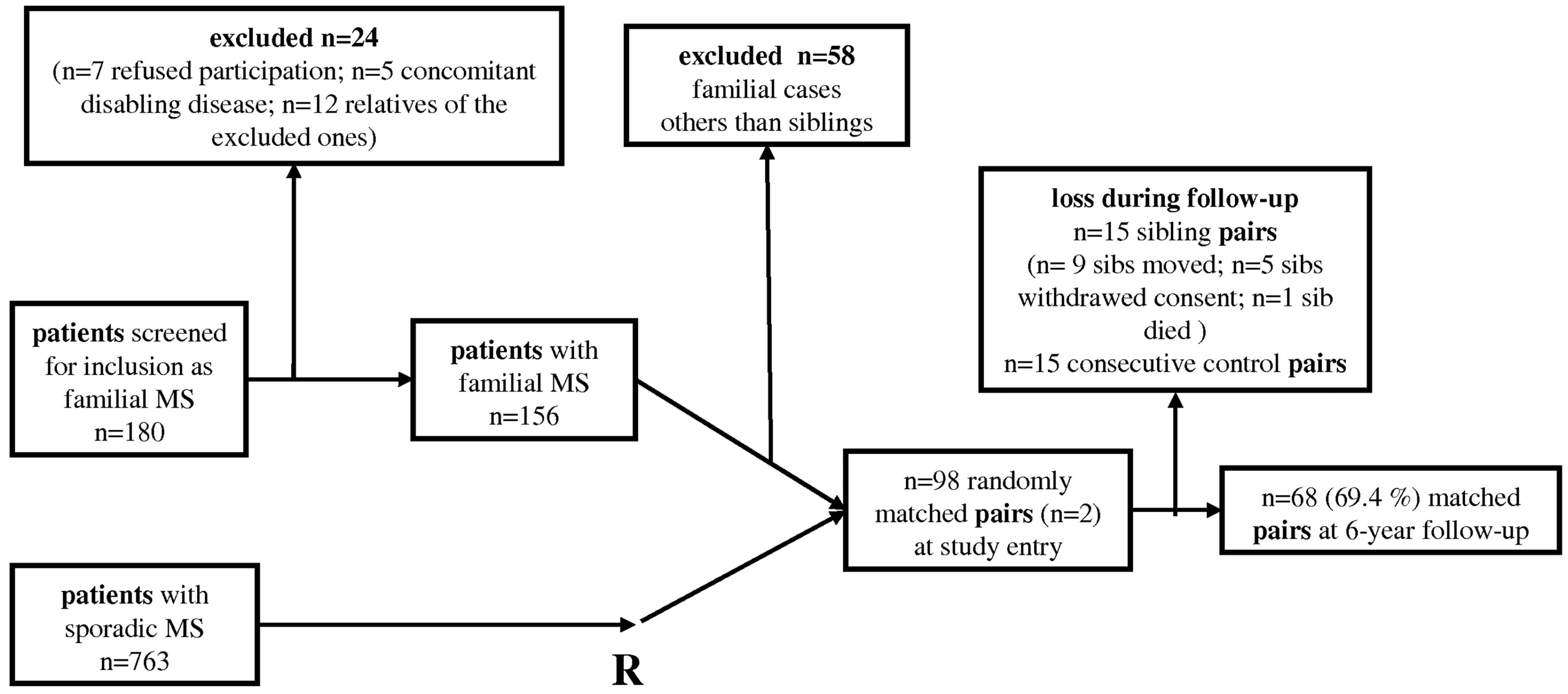
Clinical characteristic items and scores
For all siblings and their matched controls the following items were documented at study entry and continuously during the follow-up period of six years (Table 1): (1) the clinical course was defined as relapsing–remitting (RR), secondary-progressive (SP) or primary-progressive (PP); 17 (2) age at onset and disease duration were recorded from the database, external medical records or from anamnestic information; (3) any initial presentation suggestive for a first clinical event of MS, and more than 24 hours of duration, was defined as first onset and further classified as mono- or multifocal; (4) disability and involvement of functional systems were assessed using the Expanded Disability Status Scale (EDSS) (modified according to Kurtzke 18 ); (5) rate of disease progression was defined as the ratio of differences in the EDSS and the observation period in years (ΔEDSS/years); (6) conversion into secondary-progression was diagnosed after a progressive EDSS decline of ≥0.5 points over a period of at least six months without evidence of a relapse.
Data analyses
The within-group distributions of all variables of interest were analysed by means of standard descriptive techniques including graphical displays of empirical cumulative distribution functions. Most comparisons between patients suffering from familial and sporadic disease were performed using methods for matched pairs. In particular, Wilcoxon’s signed-rank statistic was used for evaluating categorical differences between familial and sporadic MS. T-tests for paired samples was performed for testing continuous variables. To test for differences with respect to the relative frequencies of two categories, we used McNemar’s test for correlated binomial proportions. The within sibling-pair association of the rates of disease progression was assessed by means of a non-parametric analogue of the intra-class correlation coefficient. Kaplan–Meier survival analysis and a hazard ratio were applied to estimate the risk for conversion into SPMS in familial versus sporadic MS. All tests were performed two-sided at the 5% level using SAS system 9.1 and SPSS 17.0
Results
Clinical characteristics
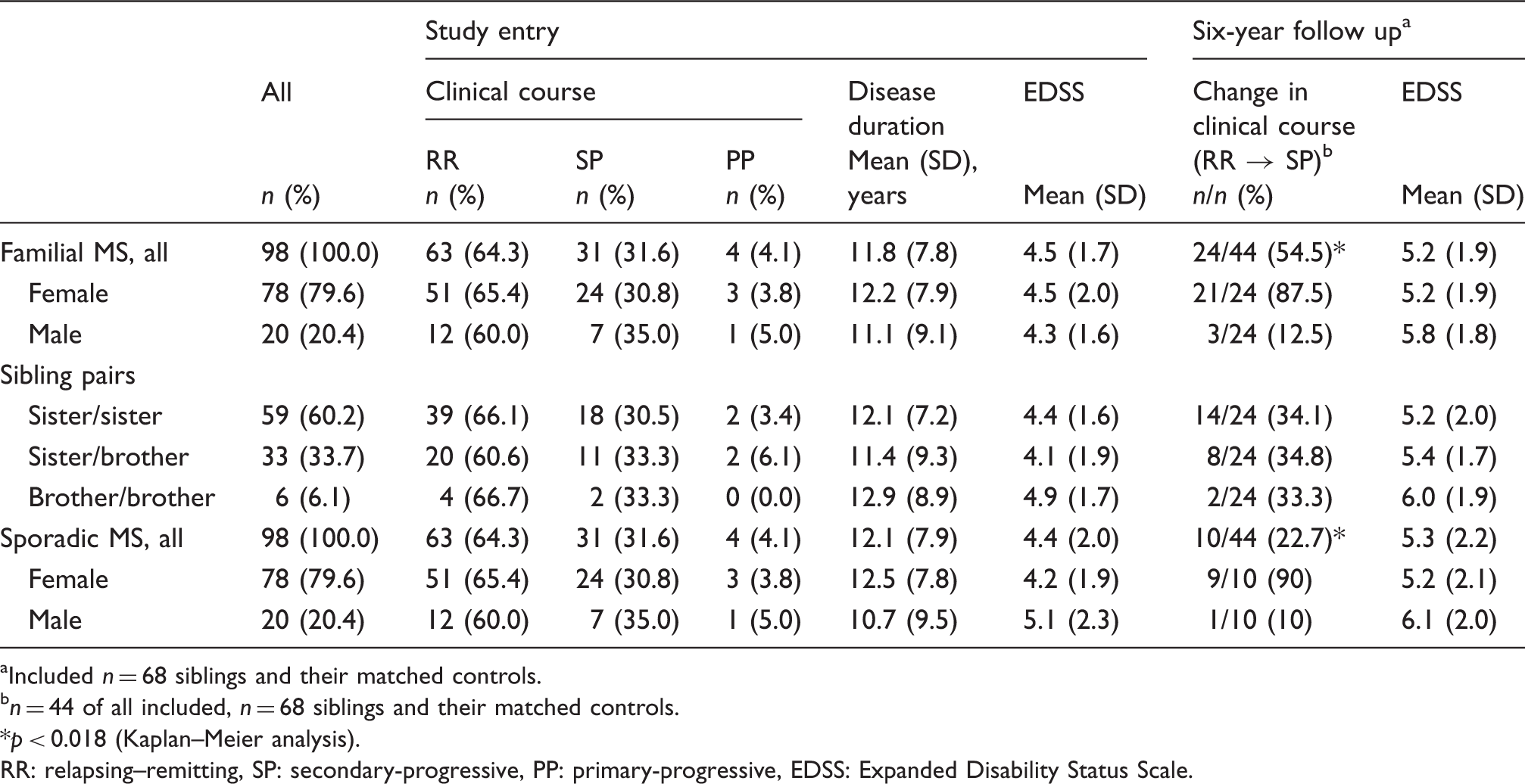
Basic statistical characteristics of the distributions of the matching criteria and the clinical characteristics of both groups are given in Table 1. The majority of siblings were sister/sister pairs (60.2%), with no obvious difference in disability or disease duration in comparison with sister/brother pairs (33.7%) or brother/brother pairs (6.1%). Mean disease duration in the familial as well as the sporadic cohort was about 12 years with an EDSS of 4.2 ± 1.7. During the six years of follow-up, the EDSS increased up to 5.2 ± 1.9 (vs. 5.3 ± 2.2 in the controls; NS). Because assessment of the EDSS is critical for analysing progression, it was not included for matching siblings with their controls. However, the pairwise distribution of differences in the EDSS between siblings and their respective controls was symmetric, with a peak at zero (not shown; p = 0.499). This suggests that both groups had an equivalent distribution in disability at study entry.
Age at onset
Figure 2 shows the empirical cumulative distribution functions of the age at onset in patients with familial and sporadic MS. The mean age at onset differed slightly but significantly between both groups (29.0 ± 9.2 familial MS vs. 29.4 ± 10.5 in sporadic cases; p = 0.0492), and was similar to epidemiologically based surveys.
19
In both cohorts, the disease onset of 80% of all patients was between 20 and 40 years; 9% occurred at age < 20 years and 11% at age > 40 years.
Cumulative percentage of age at MS onset of patients with familial (continuous line) and sporadic (dotted line) MS. Familial MS occurs significantly earlier than sporadic cases (29.0 ± 9.2 vs. 29.4 ± 10.5; p = 0.0492).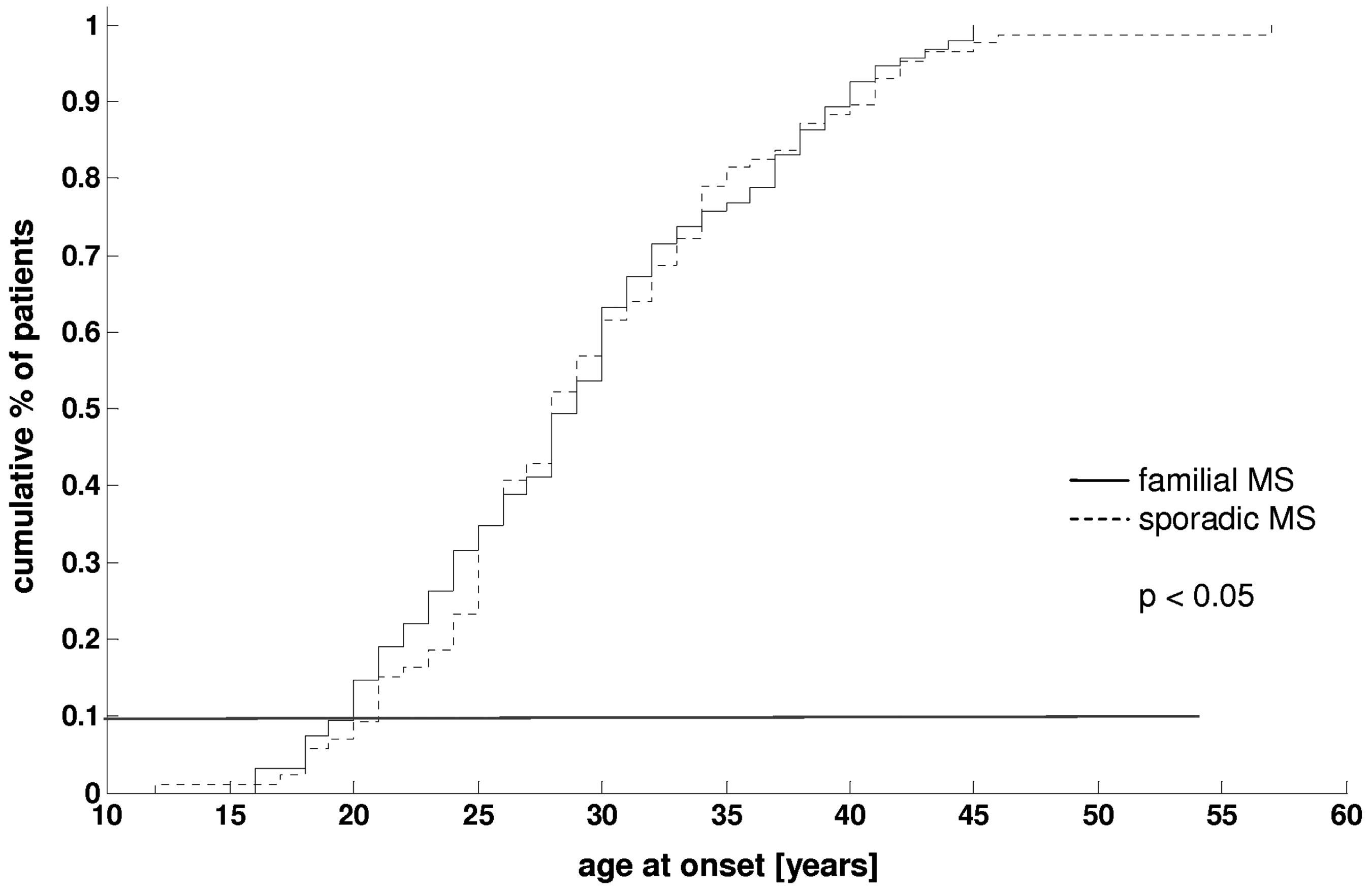
Multifocal versus monofocal onset
Clinical onset in patients with familial vs. sporadic MS

Disease progression during six-year follow-up
The distribution of the within-pair EDSS differences over the six-year follow-up period between familial and sporadic MS was symmetric, suggesting that there are no differences in disability evolution between both groups (Figure 3). Neither their means (0.158 ± 0.16 vs. 0.173 ± 0.172; p = 0.642) nor the mode of distribution differed significantly (p = 0.962). However, a significant rank correlation was obtained (r = 0.4035; p = 0.0062), after ranking the siblings within each pair according to their disease duration and after stratifying their progression into three progression risk categories: ‘low’ (EDSS ≤ 0.5/6 years), ‘medium’ (EDSS between 0.5/6 and 1.5/6) and ‘high’ (EDSS > 1.5/6). Additionally, the odds ratio for one sibling to enter the same risk category as the other was calculated to be 3.5. Both suggest that the progression rate of one appears to be predictive for the other sib.
Within-pair EDSS differences (range −4.5 to +3.5 EDSS points) over the six-year follow-up period (−4.5/6 to 3.5/6) between sib pairs and their matched paired sporadic controls. The distribution is symmetric (maximum at ‘0’), suggesting no differences between the two groups (NS; p = 0.962).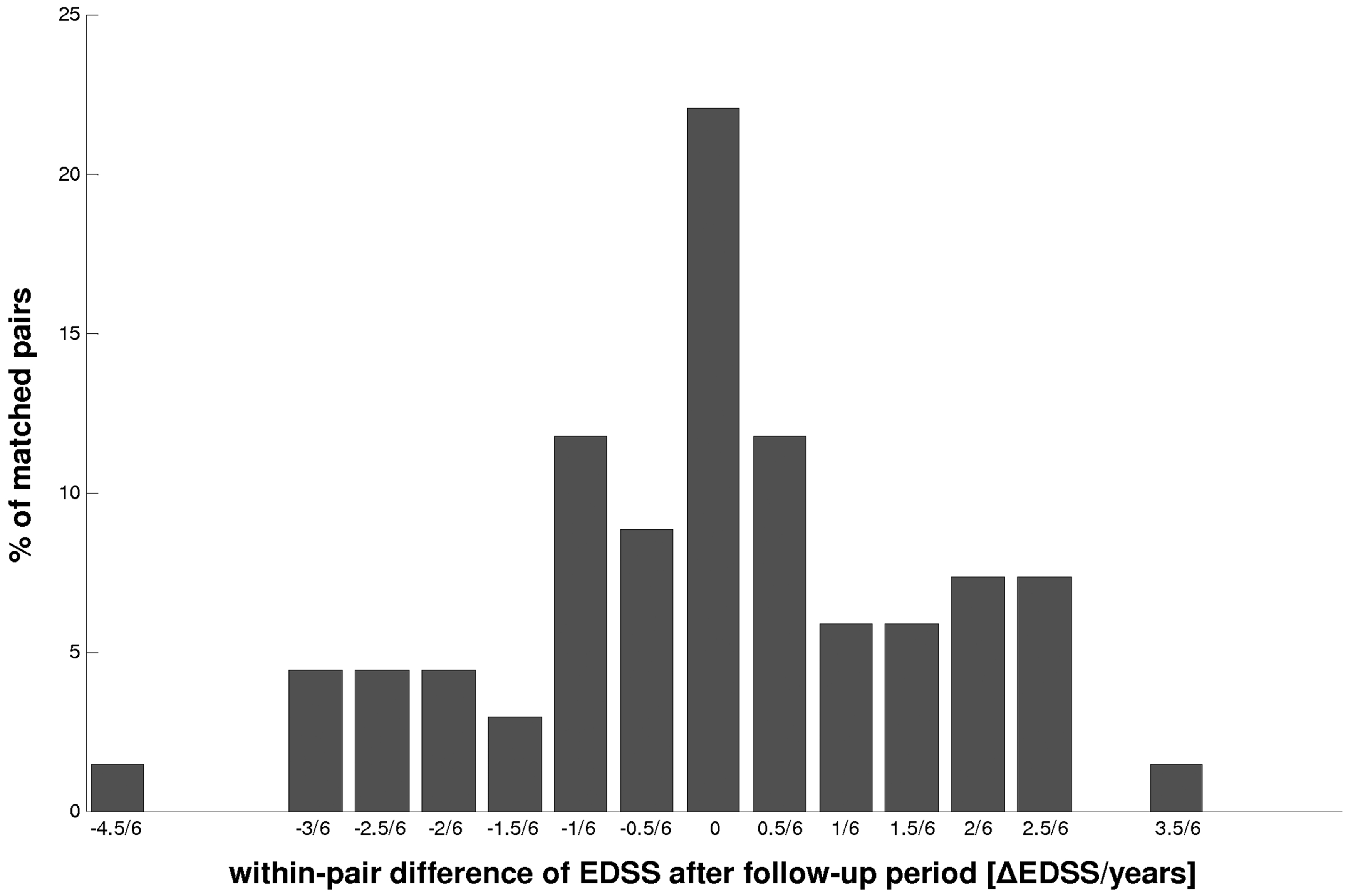
Conversion into secondary-progressive MS
From the n = 44 siblings with RRMS, who were followed over six years, n = 24 entered a secondary-progressive course (Table 1). For survival analysis (Figure 4) n = 19 patients were censored, all at the time of inclusion into this study. Though age at onset, baseline EDSS and disease duration in the subgroup of RRMS patients were similar, the odds not to convert into SPMS were significantly lower in the siblings (0.455 ± 0.075) than in their controls (0.732 ± 0.069; p = 0.018).
Kaplan–Meier analysis of the six-year conversion risk from RRMS to SPMS of n = 44 patients with initial familial RR-MS course and their respective sporadic controls. Note that during the observation period familial MS patients have a 31.8% higher risk to develop SPMS (p = 0.018).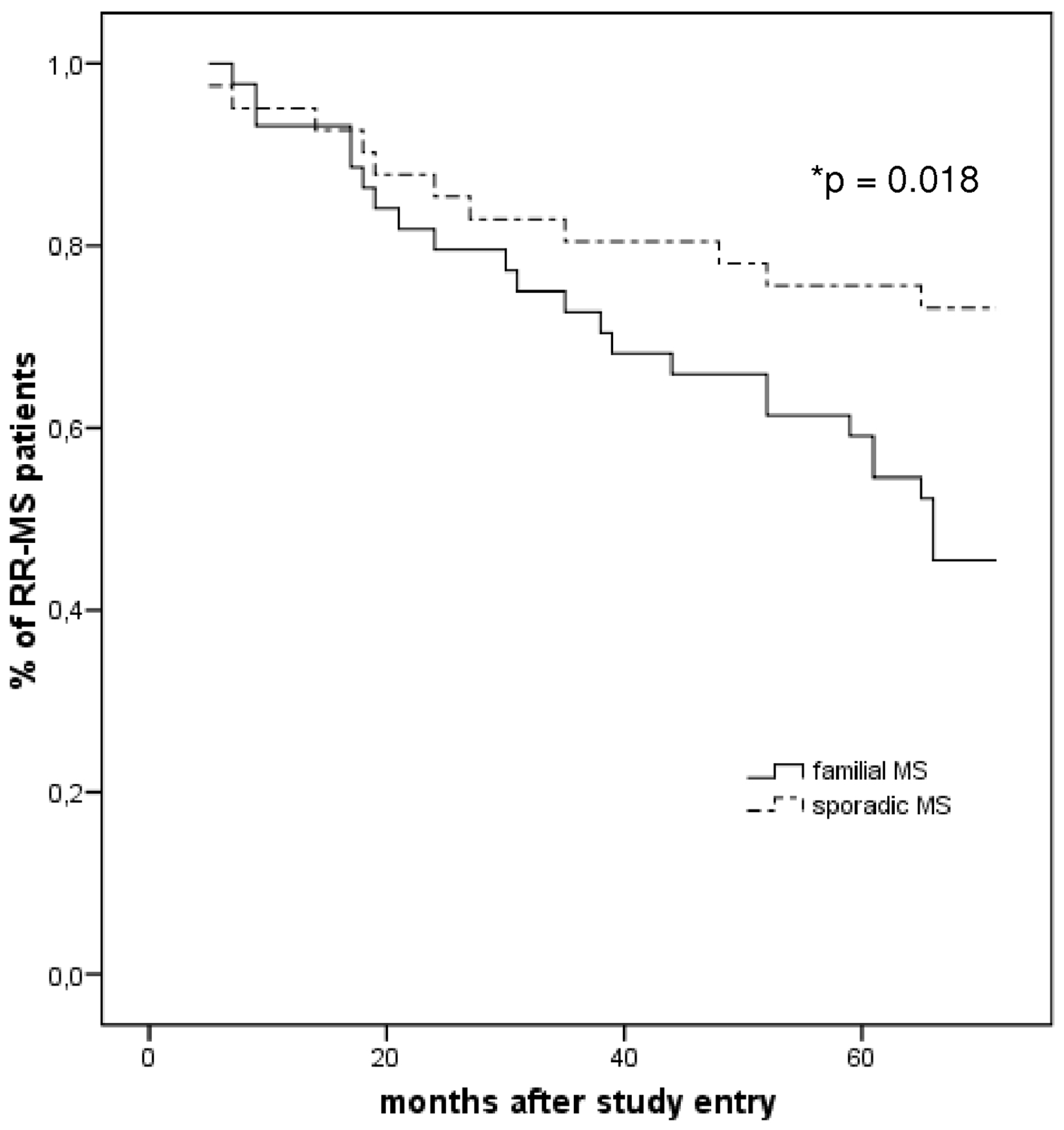
Discussion
In 7–18% of MS cases another first or second degree relative is also affected.20,21 We here provide evidence for an increased sibling risk for (1) multifocal onset, (2) within-pair correlation of progression, and (3) for conversion into secondary progression. Remarkably, key data like disease duration, baseline EDSS or age at onset in our consecutive cohort of familial cases were largely similar to those in geographically or epidemiologically based surveys.20,22 In contrast, a small Spanish cohort of familial MS patients showed lower EDSS at onset and more often a relapsing–remitting course. 23
In our prospective six-year follow-up, familial MS showed a higher incidence of progression in clinical surrogates than in sporadic cases. Secondary progression is the most common clinical course in MS, with an estimated proportion of 66% of all patients affected by MS. 22 Therefore the conversion from RR- to SPMS is a clinical and therapeutic hallmark in the course of MS. Epidemiological data suggest the evolution of disability being more dependent on disease duration and a ‘predefined schedule of progression’ than on relapses. 24 – 26 At least data from pivotal clinical trials show a higher disability evolution among SPMS patients than among RRMS.27,28 Though there is no difference in basic clinical data at study entry between our two cohorts of familial and sporadic MS, a higher risk of SPMS conversion was observed in familial cases. Beyond a constitutional finding, one possible explanation might be the sharing of MS susceptibility haplotypes responsible for the ‘schedule of progression’ among siblings in the context of epistasis. 10 Because in MS relevant epistatic control occurs in the context of distinctive HLA loci (e.g. HLA-DRB1, -DQA1, -DQB1 9 ), HLA stratified familial cohorts should be analysed for ‘immuno-disabling’ candidate genes.
However, the absence of a significant effect of familiality on the EDSS disability evolution is in line with others,29,30 suggesting a small difference which can be detected only either with larger cohorts or a longer observation period. Due to our carefully matching of sporadic pair-controls a confounding effect of biasing immunomodulatory treatment could be excluded.
Age at onset is under genetic control, 30 – 32 but the absence of significant differences between mono- and dizygotic twins suggests a rather mild influence of genes. 6 Accordingly, our patients suffering from familial MS were found to have been less than one year younger at disease onset than their otherwise matched controls. Though the clinical relevance of this finding remains elusive, early attacks are believed to be relevant not only for the initiation of but also for perpetuating MS. 12 However, genetic analyses of siblings with a focus on immunological markers of disease onset may therefore add important notes to the puzzling picture of the very early MS.
Studies focusing on clinical phenotype have revealed contradictory results.14,22,33,34 However, they conform to a heterogenous clinical presentation of MS at disease onset. Recently, the dichotomy of multifocal vs. monofocal onset became relevant for therapeutic decisions in the context of positive predictive items or disease activity assessment.12,35 Patients with familial disease presented much more often with multifocal symptoms and signs than patients with sporadic disease. Besides its prognostic relevance of familial MS in general, larger prospective studies are required to demonstrate the therapeutic relevance of these findings in familial cases.
To our knowledge, this is so far the largest population-based cross-sectional study conducted in Germany of familial MS. The reliability of the inferences made on the population underlying the samples we analysed is comparatively high. However, due to difficulties in defining the nature and extent of already known or still cryptic genes that may be involved in the MS susceptibility, there is need for larger and well characterized cohorts of familial MS patients.
Footnotes
Acknowledgments
We thank Mrs Katrin Hülsmeier, Mrs Annette Hehenkamp and Mrs Ines Jackl for excellent technical and organizational help.