
Abstract
Marburg’s variant of multiple sclerosis is a rapidly progressive and malignant form of multiple sclerosis (MS) that usually leads to severe disability or death within weeks to months without remission. Few cases have been described in the literature since the original description by Marburg. The classic pathological findings usually include highly destructive zones of extensive demyelination, necrosis with dense cellular infiltrate, and giant reactive astrocytes. We report a case of a 31-year-old woman with Marburg’s variant of MS who, over a period of eight months, became totally disabled, blind, and quadriplegic, with vocal cord paralysis, requiring a tracheostomy. The patient underwent diagnostic stereotactic brain biopsy. Clinical findings, magnetic resonance imaging (MRI), serologic and cerebrospinal fluid (CSF) findings, and neuropathology are discussed. MRI showed extensive white matter involvement in the brain and spinal cord that continuously progressed over time. A diagnostic stereotactic brain biopsy revealed extensive active demyelination with unexpected finding of active vasculitis and fibrinoid necrosis with a vascular inflammatory cell infiltrate, including polymorphonuclear neutrophils and rare eosinophils. Serologic work-up for vasculitis and neuromyelitis optica was unremarkable and the CSF showed only one oligoclonal band (OCB) not present in serum. This is the second case of Marburg’s variant of MS that demonstrated both demyelination and vasculitis. In our case these features were demonstrated simultaneously, even though the demyelination was the predominant pathological finding. Since vasculitis is not a feature of classic MS, these findings pose the question as to whether Marburg’s variant of MS is a true variant or different entity altogether.
Introduction
Multiple sclerosis (MS) is the most common among a group of diseases called ‘demyelinating diseases’ in which myelin destruction (demyelination) is the most prominent pathological feature. The profound heterogeneity in the clinical course of MS and its variants manifests itself not only in the clinical disease course, but also in the neuroradiological appearance of lesions, the neuropathological findings, involvement of susceptible gene loci, and response to therapy. This has been supported by experimental models and pathological analyses, which have demonstrated that different processes may induce MS-like demyelinating plaques, suggesting that MS may be a disease with heterogeneous pathogenic mechanisms.1–6 However, the initial heterogeneity in the earliest phase of MS lesion formation has been questioned and may disappear over time as different pathways converge in one general pattern of demyelination. 7
The basic radiological and pathological feature of MS includes multifocal demyelinating lesions (plaques) of different ages in the central nervous system (CNS). The pathological hallmarks of the lesions include a perivascular inflammatory infiltrate consisting of T cells and macrophages/microglia, a profound disturbance of blood–brain barrier in active lesions, demyelination of white matter, axonal loss, gliosis with astrocyte proliferation, remyelination especially in the early stages, and both loss and transient proliferation of oligodendrocytes.2,8–13
Other demyelinating diseases, including atypical forms of MS such as Marburg’s acute MS or Balò’s concentric sclerosis, Devic’s neuromyelits optica, Schilder’s diffuse sclerosis, and acute disseminated encephalomyelitis (ADEM) may be different in etiology and pathogenesis but share a significant part of MS pathology and show distinctive features clinically, radiologically, and pathologically.10,14–17
In 1906, Otto Marburg described three cases of a fulminant, atypical form of multiple sclerosis (MS) characterized by an aggressive course and large CNS demyelinating lesions. 18 Since that report, ‘Marburg’s variant MS’ has been used to indicate a specific form of MS distinguished mainly by the dramatic onset, rapid progression with no remission, large disseminated white matter lesions, and overwhelming neurologic deficits that result in death within weeks to months, often by destruction of vital brainstem nuclei.19–21
The neuropathological studies of cases of Marburg’s variant MS have shown: sharp-edged, highly destructive lesions or zones of extensive demyelination of almost the same age; reduction and at times complete loss of oligodendrocytes; necrosis of white matter with a dense cellular infiltration by giant astrocytes, macrophages, and microglia; relative preservation of axons; perivascular infiltration of lymphocytes and plasma cells; and reactive astrocytosis.14,15,17,19,21
A unique case report by Bitsch et al. 10 described complete lack of demyelination with infiltration of small vessel walls by mononuclear cells suggestive of vasculitis in a brain biopsy obtained from a patient 33 days after the onset of disseminated neurological symptoms. A second biopsy from the same patient 76 days later (109 days after the onset of symptoms) revealed confluent demyelinating lesions consistent with MS with preservation of vascular wall integrity within the lesion. These findings suggested a stage of lesion development prior to the onset of demyelination. Their proposal from this study was that the type of lesion found in biopsy # 1 (vasculitis) may be the so-called ‘initial’ MS lesion. 10
In this article, we report a second case that documents both vasculitis and extensive demyelination in the same patient occurring during the course of an acute, progressive demyelinating disease of the CNS that was diagnosed as Marburg’s variant MS based on the clinical course, neuroradiological, and neuropathological findings.
Case report
A 31-year-old, African-American, right-handed woman presented to the emergency department (ED) with severe abdominal pain. She was found to have urinary retention, right facial droop, and paraplegia. A few days earlier, she was complaining of leg pain, and she collapsed in the street. She was taken to another facility where an MRI showed multiple white matter lesions consistent with multiple sclerosis. The patient was given an appointment at the neurology clinic, and was discharged.
A brain MRI with and without contrast obtained in the ED showed multiple high intensity FLAIR signal lesions involving the periventricular, and subcortical deep white matter, adjacent and posterior to the fourth ventricle, and corpus callosum. Several of these lesions demonstrated nodular and ring enhancement on the post contrast imaging. Spine MRI with and without contrast showed a high STIR signal intensity lesion in the medulla, C1, C2, C6, T6–T7, and T10–T11. Several of these lesions were enhancing on the post-contrast imaging. The conus medullaris was found to be enlarged and enhancing on post-contrast imaging as well (Figure 1). Blood analysis revealed normal results including differential blood count, liver function tests, urea nitrogen, creatinine, cardiac enzymes, coagulation profile, erythrocyte sedimentation rate, C-reactive protein, vitamin B12, folic acid, and angiotensin converting enzyme level, with negative rheumatoid factor, antinuclear antibodies (ANA), C-ANCA, P-ANCA, anti-GQ1B, aquaporin-4 antibodies, Lyme titer, rapid plasma reagin and HIV screens.
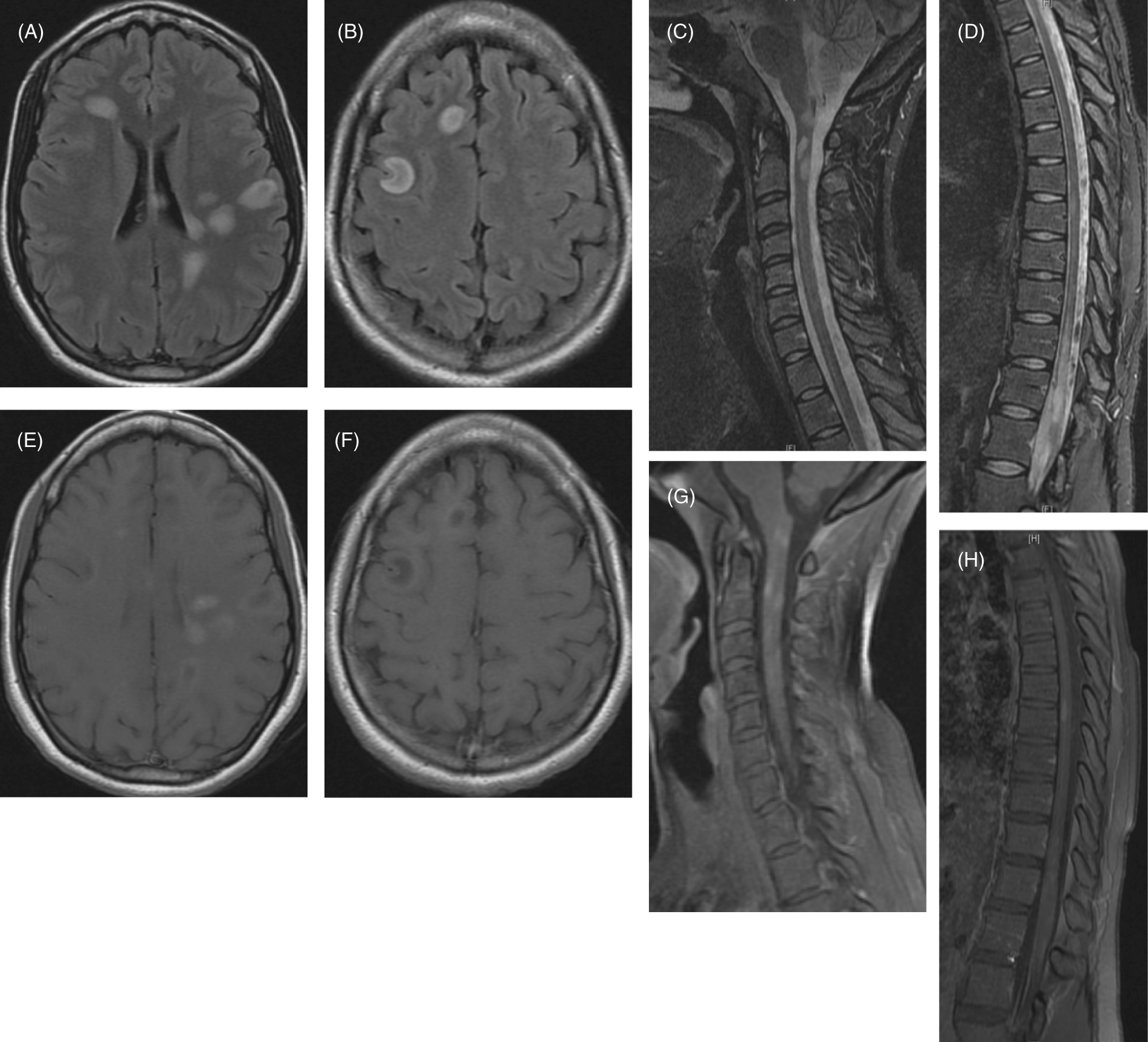
Initial MRI of the brain, cervical, thoracic, lumbar, and sacral spine. (A, B) Multiple ovoid high intensity FLAIR signal lesions involving the periventricular, subcortical, and deep white matter. (C, D, F, H) Postcontrast T1-weighted images showing multiple nodular and ring enhancing lesions. (E) STIR hyperintense lesions within the medulla and upper cervical spine with mild cord expansion at C2 level. (G) STIR hyperintense lesions at the thoracic level, the distal spinal cord and the conus medullaris, which appears to be mildly expanded.
The patient received methylprednisolone 1000 mg/day intravenously (i.v.) for five consecutive days with no response. Cyclophosphamide induction 600 mg/m2 i.v. was tried for 5 days. The patient developed leukopenia. Eventually, upon improvement of the leukopenia, the patient was discharged to a subacute rehabilitation with a follow-up planned for the neurology and urology clinics.
The patient was lost to follow-up until she was brought by her mother to the ED six months after the initial hospitalization with worsening weakness and muscle spasms. She was found to have a left gaze preference, and a right afferent papillary defect, visual acuity testing was hand motion in the right eye and 20/20 in the left eye, with a pale right optic disc. She was paraplegic with markedly increased tone, decreased sensation to light touch, pinprick, and vibration sense in bilateral lower extremities. She had mild weakness of the right upper extremity (4+/5) with right pronator drift. Deep tendon reflexes were normal and symmetric bilaterally in the upper extremities and absent bilaterally in the knees and ankles, with positive Babinski signs bilaterally. Opening pressure on lumbar puncture was 160 mm water. Cerebrospinal fluid (CSF) analysis showed a single oligoclonal band, an IgG Index: <0.6, myelin basic protein 2.4 mg/dl, glucose 90 mg/dl, protein 28 mg/dl, and no white blood cells. Brain and spine MRIs with and without contrast showed an increased size and number of T2 lesions with an increased number of enhancing lesions consistent with a progressive and active multiple sclerosis (Figure 2A–D).
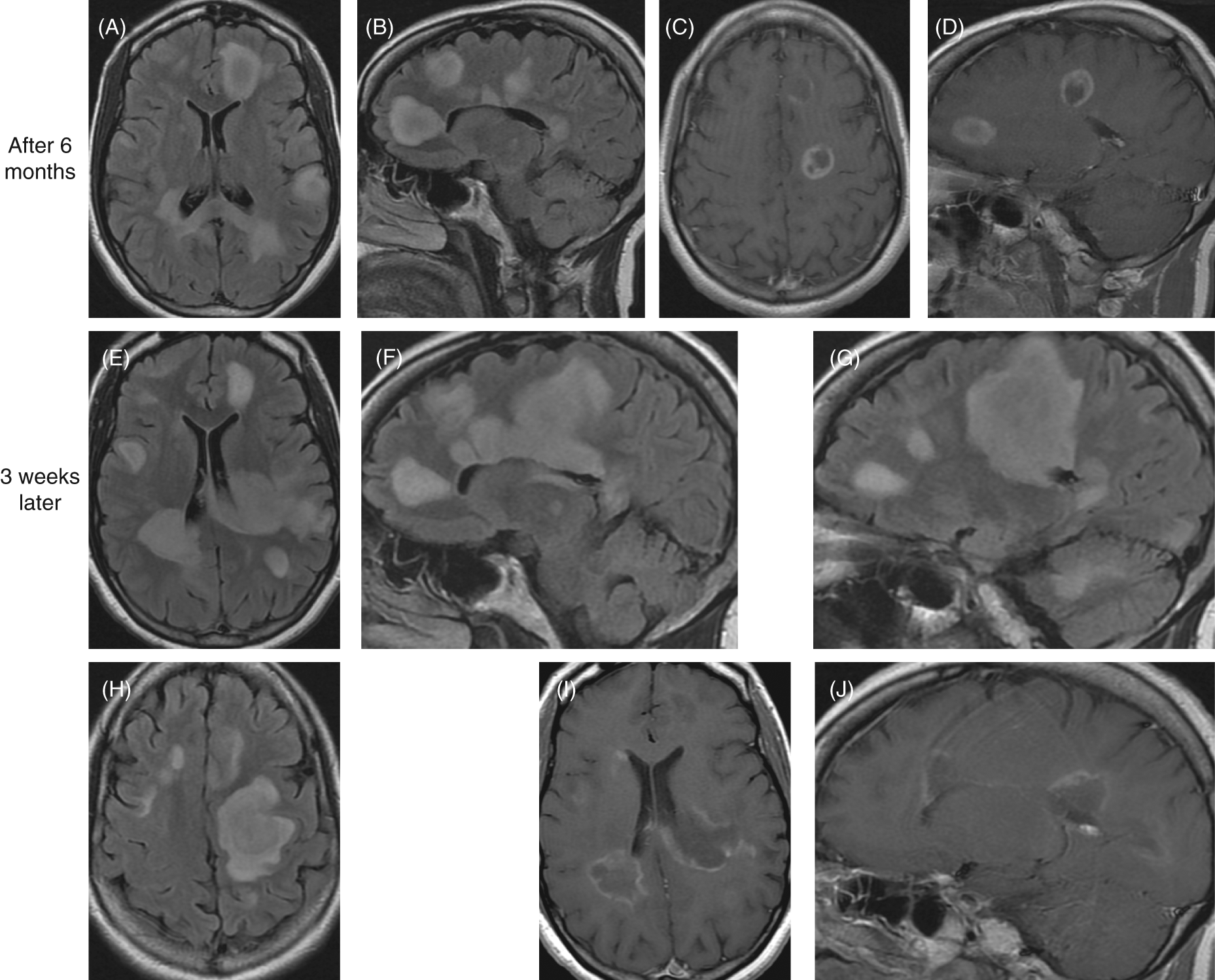
Brain MRI findings six months after initial presentation then three weeks later. Six months after initial presentation: (A) axial, (B) sagittal FLAIR showing increase number and size of the hyperintense lesions; (C) axial, (D) sagittal postcontrast T1-weighted images showing increase in size and number of enhancing lesions. Three weeks later: (E, F, G, H) increase in size and number of FLAIR hyperintense brain lesions; (I, J) postcontrast T1-weighted images showing increase in size and number of enhancing lesions.
The patient again received methylprednisolone 1000 mg/day i.v. for five consecutive days, again with no improvement. She developed a severe urinary tract infection (UTI) and was discharged to a subacute rehabilitation facility and was scheduled for a follow-up as an outpatient.
Less than three weeks later, the patient was sent to the ED from the subacute rehabilitation facility because of acute deterioration in her mental status. On examination, she opened her eyes to voice, but she was unable to follow commands. She had a dilated (6 mm) and non-reactive right pupil, with a normally reactive left pupil. There were no spontaneous movements seen in the extremities, tone was markedly increased in all four limbs, with posturing of the upper extremities and dystonic posturing of the neck. She withdrew minimally to painful stimuli. Clonus was present in both lower extremities, with spontaneous clonus noted in the right foot.
The patient was found to have a UTI and was started on antibiotics. A repeat MRI demonstrated an increase in the size and number of demyelinating lesions (Figure 2E–J). A stereotactic brain biopsy was done four days later. The biopsy was taken from an accessible gadolinium-enhancing lesion in the right frontal lobe, with tissue fixed in formalin and embedded in paraffin.
The biopsy demonstrated sharply circumscribed lesions of active demyelination with myelin breakdown products on Luxol fast blue stain, and with relative preservation of axons on Bodian silver stain, which appeared somewhat diminished in number when compared with the normal white matter, in part due to displacement by gitter cells and gemistocytic astrocytes. An antibody to neurofilament protein had an appearance that was similar to the Bodian stain, demonstrating relative preservation of axons in the completely demyelinated areas. Swollen axons were not noted with either technique. The demyelinated zones were delimited from more normal staining white matter at the edges of some of the pieces of tissues.
An unexpected finding was the presence of active vasculitis, with fibrinoid necrosis of some blood vessels in the specimen, with a mixed vascular inflammatory cell infiltrate including polymorphonuclear neutrophils, mononuclear cells, eosinophils, and rare plasma cells (Figure 3), best seen in demyelinated zones. Microabscesses were not observed. On immunohistochemistry with an antibody to cluster of differentiation (CD)8 T cells there were several positively stained cells in the perivascular cell infiltrates, as well as scattered, single cells throughout the abnormal white matter. An antibody to CD4 T cells also stained some cells in the perivascular infiltrates, with only rare CD4-positive cells in the abnormal white matter. An antibody to the B-cell marker CD20 stained only rare cells in the white matter.
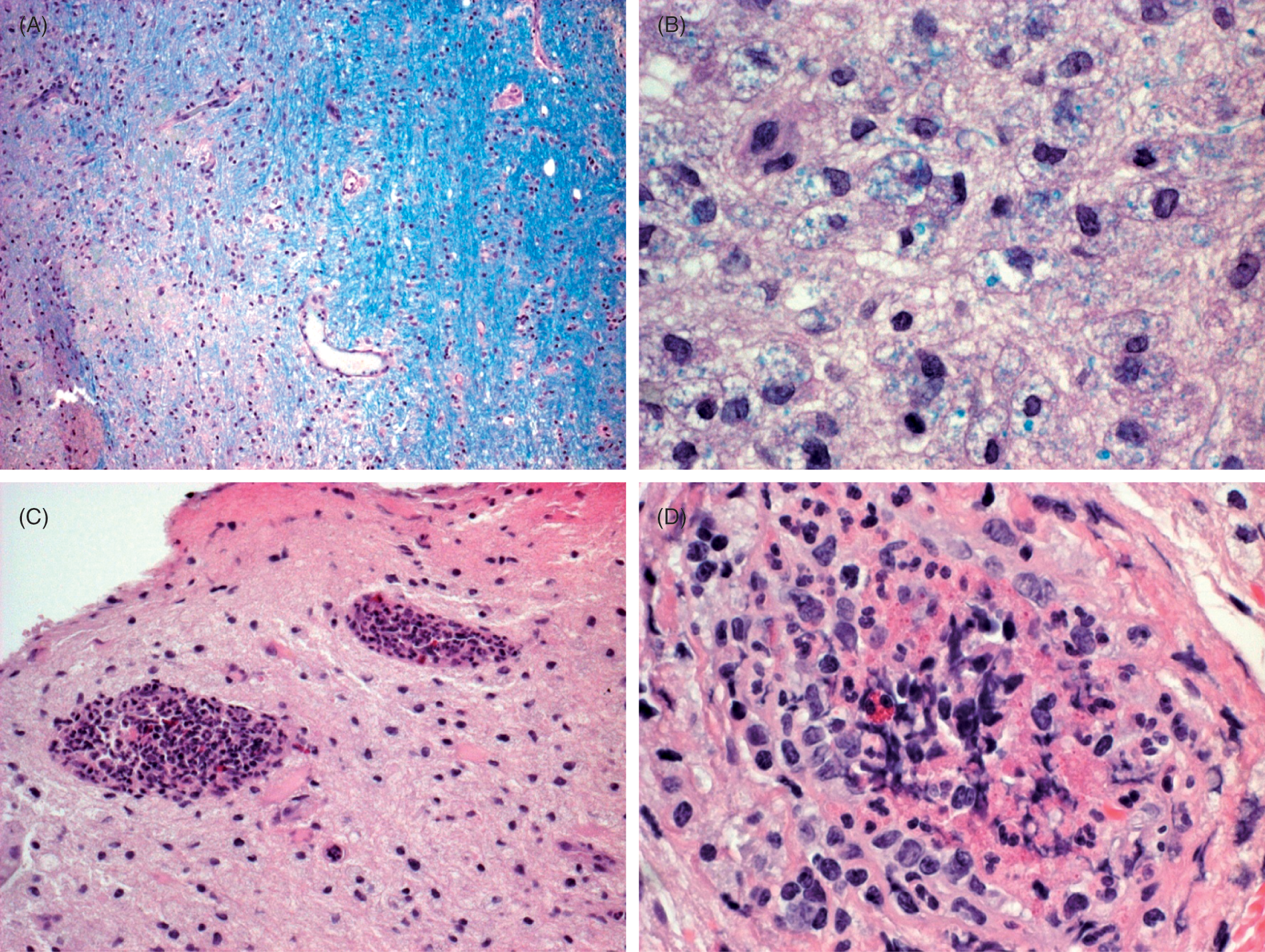
Unexpected findings of the brain biopsy showing vasculitis and extensive demyelination. (A) Edge of plaque, with normal myelin on right and demyelinated white matter on left. Luxol fast blue -periodic acid Schiff (LFB-PAS) stain, original magnification ×10. (B) Demyelinated region with macrophages containing LFB-positive material, indicating recent demyelination. LFB-PAS stain, original magnification ×25. (C) Vasculitis involving two vessels in demyelinated region of white matter. Eosinophils are present in the inflammatory infiltrates. Hematoxylin and eosin (H&E) stain, original magnification ×10. 3D: Vasculitis in demyelinated region, with fibrinoid necrosis. Note an eosinophil. H&E stain, original magnification ×25. (Colour online only)
The patient received 1 gm/m2 of cyclophosphamide i.v. with no improvement. A few days later, she developed respiratory stridor, and was found to have vocal cord paralysis that necessitated an emergent endotracheal intubation and subsequent tracheostomy. She was transferred to the medical intensive care unit. She developed neutropenia and sepsis, and lost blink to threat. She did not improve and was transferred to a nursing home with arrangements for follow-up.
At the last follow-up visit one month later, it was noted that the patient had not had any further relapses or progression of her MS, but she had developed focal seizures. Her neurological examination was unchanged except for some improvement in vision in the left eye.
Discussion
The patient in this case report presented with a rapidly progressive neurologic disease with extensive dissemination of demyelinating lesions. Clinical and radiological data were suggestive of a fulminant demyelinating disease, and the patient was diagnosed with acute MS of Marburg type. The diagnosis was supported by the absence of knee jerks that suggested peripheral nervous system involvement, 14 lack of multiple oligoclonal bands on CSF examination,10,17,19,22 continuous progression of MRI lesions, including size and number of T2 and T1 enhancing lesions over time, consistent with progressive and active multiple sclerosis.17,20,23. Neuropathological findings confirmed the presence of active demyelination.
Patients with the Marburg’s variant of MS typically have a poor prognosis, often with a fatal outcome. However, the availability of intensive care units has improved the prognosis/survival considerably over the past few years, with cases reported in the literature that survived and reached clinical stability, albeit with severe residual disabilities.20,24–26
The differential diagnosis of fulminant demyelinating diseases includes MS of Marburg’s type, Balò’s concentric sclerosis, Schilder’s diffuse sclerosis, and acute disseminated encephalomyelitis (ADEM). All these subtypes share a significant part of MS pathology; however, each of them harbors discriminative features clinically, radiologically, as well as pathologically.11,17,23,27.
Balò’s concentric sclerosis is characterized pathologically and radiologically by alternating layers of myelin preservation or remyelination and myelin loss that can be seen radiographically on T2-weighted and contrast-enhancing T1-weighted MRI images as a distinctive pattern of concentric rings or whorled appearance of hypo/isointense and hyperintense rings,11,14,15,28–30 features not seen in the case under discussion.
Schilder’s diffuse sclerosis is a very rare disease of different etiologies affecting mainly children, and it shows a response to corticosteroid therapy. Clinical presentation and radiological findings may mimic a tumor or abscess. 31 - 34 Poser et al. 35 in 1986 suggested criteria that can distinguish this diagnosis, which needs to be differentiated from various leukodystrophies. It is a subacute or chronic disorder resulting in the formation of one, or more commonly two, roughly symmetrical bilateral plaques measuring at least 3 × 2 cm involving the centrum semiovale of the cerebral hemispheres with no other lesions detected either clinically, paraclinically (e.g. by visual evoked potential), or radiologically, with no involvement of the peripheral nervous system, normal adrenal functions, and normal very long-chain fatty acids. 35
ADEM is another rare and acute demyelinating disorder, often with a favorable long-term prognosis. Maximal deficits can be reached within several days and remission can be similarly rapid. ADEM is thought to be an autoimmune response to myelin basic protein that is triggered by infection or immunization, occurring predominantly in children and young adults. One of the proposed criteria to differentiate ADEM from other forms of MS is gray matter involvement.22,36,37 Pathologically, inflammation and demyelination in ADEM lesions are predominantly perivenous. ADEM lesions would be expected to have a stable appearance on MRI with no new lesions on long-term follow up MRIs.11,38,39 By contrast, the patient in this report lacked a known history of exposure to vaccines or infections; demyelination was diffuse in white matter and was not confined to the tissue around inflamed vessels, and most important, the patient had had progressive and active disease for several months.
Other diagnoses that were considered, such as primary CNS lymphoma, CNS vasculitis, and progressive multifocal leukoencephalopathy, were excluded by the biopsy results. Also, perivascular eosinophils have been reported in documented cases of neuromyelitis optica (NMO). 40 However, NMO was ruled out clinically when aquaporin-4 antibodies were found to be negative in this patient. In sum, the clinical, radiological, serologic, and pathological findings in our patient did not justify any diagnosis other than acute MS of the Marburg type.
A striking finding in this patient was the brain biopsy that was done almost 8 months after the onset of neurologic symptoms, with demonstration of active demyelination with myelin breakdown products, and with relative preservation of axons. An unexpected finding was the presence of active vasculitis with fibrinoid necrosis and a perivascular mixed cell infiltrate, including a small number of eosinophils.
While it is possible that this patient had both vasculitis and demyelination from two different diseases processes, or a primary CNS vasculitis, there are several lines of evidence suggesting that this is unlikely. No evidence of a vasculitis was found on extensive laboratory testing and there was no clinical evidence of vasculitis in other organs including the eyes. Further, the pathology showed primarily extensive demyelination with sparing of axons. While sepsis can worsen MS, it would be unlikely to cause vasculitis with absence of microabscesses in the pathology. Most importantly, this patient did not have any signs of sepsis, or evidence of sepsis on the multiple blood cultures done prior to brain biopsy.
The literature contains few case reports of Marburg’s variant of MS with biopsy or autopsy findings17,19,21,27, and only one, by Bitsch et al., 10 describing both demyelination and active vasculitis in a single patient. This patient was diagnosed with Marburg’s variant of acute MS and underwent two diagnostic stereotactic brain biopsies from different lesions at different sites on day 33 and 109 after the onset of the neurologic symptoms. The first biopsy showed no demyelination, but active vasculitis. However, the second biopsy showed active confluent demyelinating lesions with preserved integrity of the vessel walls. Bitsch and colleagues proposed that the type of lesion found in the first biopsy could be the so-called ‘initial’ MS lesion. Based on their findings, they suggested the following features to be possibly contributing to the early MS lesion:(1) mononuclear cells migrating through small CNS blood vessels, (2) a diffuse inflammatory infiltrate of the white matter, (3) lack of demyelination, (4) preservation of oligodendrocytes, and (5) early expression of toxic mediators such as tumor necrosis factor α and nitric oxide (NO).
The appearance of vasculitic changes in the setting of widespread classic demyelination in our patient confirms the findings of Bitsch et al., even though in our patient these features were detected simultaneously. If Bitsch’s theory is correct, the occurrence of both vasculitis and demyelination at the same time could be explained by the different age of the lesions. Vasculitis, which may be focal, can easily be missed on biopsies due to sampling.
Further pathological studies are needed to confirm whether the presence of CNS vasculitis in patients with Marburg’s variant of MS is a part of the ‘initial’ MS lesion and whether the speed of development and progression of lesions may often be too rapid to allow it to be seen. It is not clear whether CNS vasculitis could be a part of the pathology of Marburg’s variant of MS. If this is the case, Marburg’s variant might not be a true variant of MS but a different entity altogether.
Understanding the diverse immunological and pathological mechanisms that lead to the development of CNS lesions in different forms of MS and MS variants, as well as the timing of these various processes, is crucial for designing effective therapies and optimal therapeutic strategies that can target certain pathways or mechanisms at specific times during the course of the disease.
Because Marburg’s variant MS is rare, no controlled clinical trials have been done, and no therapeutic intervention has been consistently successful.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.